![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
30 Cards in this Set
- Front
- Back
|
Wernicke-Korsakoff Syndrome |
-Mutation in the transketolase gene - Decreased affinity to TPP |
|
|
Farber disease (disseminated lipogranulomatosis) |
- Enzyme deficiency resulting in accumulation of PAS-positive lipid consisting of ceramide, leading to cell damge with inflammatory response |
|
|
Phenylketonuria |
- Phenylalanine hydroxylase defect |
|
|
Maple syrup urine disease |
- Branched alfa-ketoacid dehydrogenase complex - Defective metabolism of branched chain amino acids (Leu, Ile, Val), accumulation of keto acid |
|
|
Tyrosinemia I |
Fumaryl-acetoacetate hydrolase deficiency |
|
|
Tyrsinemia II |
Tyrosine aminotransferase deficiency |
|
|
Citrullinemia type I |
Argininosuccinate synthetase deficiency |
|
|
Homocystinuria |
Cystathionine B sythase deficiency |
|
|
Methylmalonic acidemia |
Glycin and methylmalonic acid accumulates. Can depend on cofactor B12 - Methylmalonyl-CoA mutase inhibited |
|
|
Propionic acidemia (propionemia) |
Lycine, propionate and "strange" ketonbodies accumulates. - Propionyl-CoA carboxylase inhibited |
|
|
Galactosemia |
Gal-1-P uridyl transferase or galactokinase or UDP-galactose-4-epimerase deficiency - Gal-1-P accumulating in liver will give liver chirrhosis |
|
|
Fructosuria |
Deficiency of hepatic fructokinase enzyme |
|
|
Fructose intolerance (Frucosaemia) |
Fructose-1-P aldolase enzyme deficiency -> inhibition of glycolysis, glycogenolysis, and gluconeogenesis. |
|
|
von Gierke's disease |
Glucose-6-phosphatase deficiency -> hepatomegaly becasue of glycogen accumulation |
|
|
Pompe's disease |
a-1,4-glucosidase enzyme, lysosomal glycogen storage diseases |
|
|
Cori's disease |
Amylo-1,6-glucosidase (debranching enzyme) -> glycogen accumulation in the liver, heart and skeletal muscle |
|
|
McArdle's disease |
Glycogen phosphorlyase deficiency in the muscle |
|
|
Glucose-6-phosphate dehydrogenase deficiency |
X-linked dominant oxidative stress->hemolysis-> anaemia Protection against malaria G6PD / NADPH pathway is the only source fro reduced glutathione in RBS, which protects against oxidative stress. In case of deficiency, denatured, precipitated haemoglobin aggregates to form Heinz bodies |
|
|
Alkaptonuria |
homogentisate 1,2-dioxygenase enzyme deficiency |
|
|
GM1 gangliosidoses |
B-galactosidase deficiency -> abnormal storage of acidic lipid materials in PNS and CNS, particularly in nerve cells |
|
|
Tay-Sachs disease |
Hexosaminidase A deficiency - Nomally a hyrdolytic enzyme found in lysosomes, which breaks down phospholipids. - Hyrdolysis of GM2-ganglioside requires 3 enzymes, two of which are hexosaminidase A subunits, the third is an activator. -GM2-ganglioside accumulates in neurons |
|
|
Sandhoff disease |
B-hexosaminidase B deficincy -> progressive destruction of CNS |
|
|
Gaucher's disease |
Glucocerebrosidase enzyme deficiency. -> glucocerebroside accumulates, Gaucher cells appears |
|
|
Niemann-Pick disease |
Sphyngomyeline phosphodiesterase deficiency (sphyngomyelinase -> accumulation of sphyngomyeline in spleen, liver, lungs, bone marow and brain |
|
|
Hyperglycinemia |
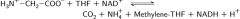
Defect of glycine cleavage enzyme: |
|
|
Tyrosinemia III |
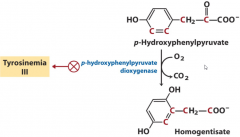
|
|
|
Lesch-Nyhan syndrome |

|
|
|
Congenital hyperammonia type I |
Carbamoyl phosphate synthetase |
|
|
Congenital hyperammonia type II |
Ornithine transcarbamoylase |
|
|
Albinism |
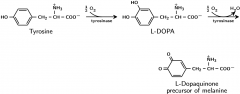
|