Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
103 Cards in this Set
- Front
- Back
|
-What is the approximate VDW radius of water?
-What is the bond angle of water? |
-1.2-1.4 A
-104.5 deg |
|
|
Define pK
Why is this useful for buffering? |
pK of an acid is numerically equal to the pH of the solution when the molar concentrations of the acid [HA] and its conjugate base [A-] are equal
- A solution at a pH near the pK of that specific solution is relatively resistant to pH changes and is a good buffer |
|
|
Define zwitterion.
Why does acetic acid raise the pK1 of amino acids? |
-molecules that bear charged groups of opposite polarity
-replaces an amino group with a methyl group; removes the zwitterionic state of an AA, making it harder to deprotonate the OH group |
|
|
How can you determine if an AA is L- or D?
|
- Perform polarized light studies
-If you have the structure in front of you, can spell "CORN" if you go clockwise around the molecule (counterclockwise = D) while looking at the H atom substituent. |
|
|
Define enantiomer.
Define chirality. |
Molecules that are nonsuperimposable mirror images of one another, and can only be distinguished by probing with plane-polarized light or other reactants that contain chiral centers.
Chiral Carbons are attached to 4 different substitutents. |
|
|
What AA is not optically active? Why?
|
Glycine, because it is not chiral.
|
|
|
What are stereoisomers?
|
-A molecule may have multiple asymmetric centers
-Stereoisomers are molecules with different configurations about one chiral center -Molecule with n chiral centers has 2^n different possible stereoisomers and 2^n-1 enantiomeric pairs |
|
|
Define diastereomers.
|
Optical isomers of L- and D- AA which have more than one chrial center, AKA "allo" forms.
-The allo forms have the same chemical makeup, but different structure and chemical properties |
|
|
What are torsion angles?
|
-Define the position between 4 atoms (A, X, Y, and B) joined by 3 bonds (Newman projections)
-cis = 0, trans = 180 -(+)= rotate to right -(-)= rotate to left |
|
|
-How are torsion angles used to describe AA?
-What is the X (chi) value? -What is a rotamer? |

.
|
|
|
What are the non-covalent types of interactions observed in proteins?
|
-Van der Waals forces
-Hydrogen bonds -electrostatic interactions (salt bridges) -Hydrophobic forces |
|
|
Van Der Waals forces
|
-weak packing forces between all protein atoms
-atoms have electron clouds which can't interact, yet the atoms need to be close enough to interact with one another -occur between electrically neutral molecules -arise from permanent and/or induced dipoles - can be divided into 3 subtypes: -dipole/dipole -dipole-induced dipole -London dispersion forces |
|
|
How do the different types of VDW forces vary?
|
-dipole/dipole: 2 permanent dipoles interact
-dipole-induced dipole: strong permanent dipole induces a weak dipole in a neutral molecule -London dispersion forces: random e- movements cause a charge imbalance which results in inducible dipoles between nearby molecules |
|
|
Describe non-covalent electrostatic interactions.
|
-AKA ion pair or salt bridge
-are usually highly solvated -NOT directionally dependent -can influence protein interactions from 2.8-10 A |
|
|
Define dielectric constant.
|
D: relative dielectric constant of a material under given conditions is a measure of the extent to which it concentrates electrostatic lines of flux
-water has a D of 78.5, benzene around 2.3 |
|
|
What is Coulomb's Law?
|
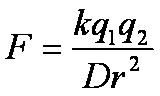
k= constant
D= dielectric constant r= radius that separates two electrical charges q1/q2= electrical charges 1 and 2 F= force between 2 electrical charges |
|
|
-What is the average distance for an H bond?
-Is the distance associated with strength? -How do these bonds differ from salt bridges? |
-2.5-3.5A; usually around 2.8A
-distance can be associated with strength -H bonds are directional |
|
|
What AA are normally (-) at physiologic pH? (+)?
|
(-): asp, glu
(+): his, lys, arg |
|
|
-Describe the peptide bond.
|
- describes the bond between the carbonyl C and amide N
- the bond has characteristics of a double bond due the lone pair e- on the N - causes a dipole, making the O slightly (-) and the N (+) - due to the partial double bond characteristic, this bond is planar |
|
|
The angle about the C(a)-N bond is ________, while the angle about the C(a)-C(o) bond is _________. What values are not allowed for these angles?
|
- Phi ("piecan" = Phi-CaN)
- Psi ("pyschic" = Psi-CaC) -Phi 180, Psi 0 due to overlap of N-H hydrogens and Phi 0, Psi 180 due to overlap of carbonyl oxygens |
|
|
In a Ramachandran plot, what quadrants and Psi/Phi values are typical for:
-antiparallel B sheet -parallel B sheet -Left-handed helix -Right-handed alpha helix |
- upper left, Psi ~ 130, Phi ~-130
-upper left, Psi ~ 90, Phi ~115 -upper right, Psi 45, Phi 45 -lower left, Psi -50, Phi -60 |
|
|
What quadrant has very few probable structures? Why? What is the exception?
|
- the Lower Right hand quadrant, because steric hindrance and electron cloud overlap of the atoms prevents this "cis" type peptide chain formation
-The exception would be for a compound which contains a lot of glycine |
|
|
What are the organizational levels of protein structure?
|
-AA
-primary structure (linear AA chain) -secondary structure (alpha helices and beta sheets) -super-secondary structure (AKA motifs) -tertiary structure (relationship of secondary structure in 3-D space; Domains) -quaternary structure (spatial arrangement of more than one protein chain) |
|
|
The peptide backbone is characterized by what three torsion angles?
|
-phi: Ca-N bond
-psi: Ca-C bond -omega: describes each Ca on either side of the C-N bond; is almost always 180 deg (trans) |
|
|
What is special about proline and torsion angles?
|
- Proline HAS to have a Phi of -60 since the R group is covalently bonded to the amide N
|
|
|
Alpha helix:
Each AA extends ____ angstroms (A) along the helix axis, with _____ residues/turn. Every R group is separated by ____ degrees. |
-1.5 angstroms
-3.6 residues/turn -100 degrees |
|
|
-Each peptide carbonyl in a alpha helix participates in what?
-Are alpha helices polar? Why or why not? |
-is hydrogen bonded to the peptide N-H group 4 residues further up the chain.
NOTE: the first 4 amide N's and last 4 carbonyl O's cannot participate in H-bonding - Yes, because there are net dipole movements due to the (-) carbonyl O and (+) amide N |
|
|
How does a 3(10) helix differ from an alpha helix?
|
- fewer residues/turn (only 3 as opposed to 3.6)
- H-bonding occurs only 3 residues up the chain (as opposed to 4) |
|
|
-Why are left-handed helices made of L-AA unfavorable?
-On a Ramachandran plot, what are the X and Y axes? |
- because the close contact of the R group with the carbonyl group
-X: Phi; Y: Psi |
|
|
In Beta sheets, what are the directions of the AA groups?
|
- If the C-N peptide bond is in line with the plane of a sheet of paper; the N-H and carbonyl O would jut out at 90 deg angles parallel to the sheet, and the R groups would be coming out of the page; alternating top and bottom
|
|
|
Secondary structures change direction; ______ are 4 AA-long and reverse the direction of the AA chain while _______ can be any length.
|
-beta turns (AKA reverse turns) are 4 AA long and require -XPGX-
-loops can be any length |
|
|
What are the torsion angles of a beta turn?
|
There are three types:
Type I: phi2 -60; psi2 -30; phi3 -90; psi3 0 Type II: phi2 -60; psi2 -120; phi 3 90; psi3 0 3(10) helix: no glycine; Phi2 -60; psi2 -30 |
|
|
What is the difference between type I and II beta turns?
|
the only difference is the flip in position of the carbonyl oxygen
|
|
|
Define MOTIFS.
|
Motifs, AKA super-secondary structures, are small pieces of connected secondary structure that are repeated in protein structures.
They are often required for the structural integrity of the protein but are not stable by themselves. There are helical and beta motifs |
|
|
What are the different types of MOTIFS?
|
Helical:
- alpha/alpha: structural - helix-turn-helix: functional - EF hand: functional Beta: - beta hair pin: structural - beta/alpha/beta: structural |
|
|
Describe the different helical motifs.
|
- alpha/alpha: structural; used to build 4-helix bundle domains; reverses direction of an alpha helix
- helix-turn-helix: functional; DNA binding proteins - EF hand: functional; binding Ca ions |
|
|
Describe the different beta motifs.
|
-beta hairpin motif: structural, links two anti-parallel beta strands
-beta/alpha/beta: structural, links 2 parallel beta strands; adopts a right-handed superhelix and is the building block of many enzyme structures |
|
|
What are the types of connections between beta strands?
|
- hairpins: connect adjacent antiparallel beta strands
- cross-overs: connect adjacent parallel beta strands; often site of beta/alpha/beta loop |
|
|
What are protein domains?
|
-3-D folding units that occur w/in one polypeptide chain
-usually contain a hydrophobic core -may or may not be stable away from the full length protein -binding or enzyme sites are often found between two domains -allow for conformational changes to occur during substrate binding -multiple domains allow the segregation of protein function |
|
|
What are the types of protein symmetry?
|
-rotational (cyclic): degrees of rotation to arrive at the equivalent point (360/subunit)
-dihedral: 2 perpendicular 2-fold rotations axes that result in 3 perpendicular twofold axes |
|
|
Do proteins have mirror symmetry? Why or why not?
|
No, because to get a direct mirror image (instead of a rotational image) would require use of both D and L AA
|
|
|
What is the Van der Waals contact distance?
|
-the most favorable distance when it comes to two atoms and the electrical potential of London dispersion forces
-the optimal non-bonded non-hydrogen bond contact distance for any atoms is the sum of the two atoms VDW radii |
|
|
What are the differnt types of protein surfaces?
|
-VDW surface: represents the volume of the atoms
-Molecular (contact) surface: surface generated by contact with a probe such as water that is represented by a sphere with a VDW radius of 1.4 A -Accessible surface: most often cited; defined from the center of the molecular probe described for the molecular surface ("water accessible") |
|
|
What are types of protein motion?
|
-atomic fluctuations: vibrations of individual bonds and atoms
-collective motions: groups of atoms from one sidechain to an entire domain undergo continual motion -triggered conformational changes: groups of atoms move in response to a stimulus; this motion is now being challenged |
|
|
What are the ranges, in angstroms, of the types of protein motion?
|
- atomic fluctuations: 0.01-1 A
-collective motion: 0.1-10 A -triggered conformational changes: varies |
|
|
What are atomic fluctuations described by?
|
temperature factors or B-factors in X-ray crystal structures
|
|
|
What are the two models for how protein motion regulates binding and function?
|
-classical ligand: ligand binds to protein and induces conformational change and subsequent catalysis
-fluctuated conformations: all possible conformations of a protein exist, even when a ligand is absent, and the "unbound" conformation is dominant unless ligand binds, resulting in a shift towards the active state (supported by studies that show PO4-alation alter protein flexibility) |
|
|
Describe the following molecular dimensions:
-angstrom (A)= -H-bond: -C-C bond: -11 residue alpha helix: -11 residue beta strand: |
-10^-10 meters
-2.8 A -1.5 A -15 A long (1.5 A/residue) -35 A long (3.5 A/residue) |
|
|
How is the average distance between the backbones of superimposed proteins measured?
|
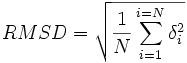
.
|
|
|
What are some properties of protein crystals?
|
-are 40-90% water
-contain large solvent channels that allow small molecules to diffuse into the crystals -does not alter the shape of the protein tertiary or quatenary structure |
|
|
What is the difference between a unit cell and an asymmetric unit?
|
-UNIT CELL: smallest volume of a protein crystal that can be repeated or "stacked" in 3-D to generate a crystal
-Asymmetric unit: the smallest volume of a unit cell that can be generated to make that unit cell (can be found in the PDB file) |
|
|
What do crystal structures represent?
|
-the average structure of all molecules that make up the crystals
|
|
|
-Reactions that are coupled can combine the _______ but not the ____.
-How much energy is required to substantially shift the K value? |
-delta G (Gibbs Free Energy)
-K -not large (due to natural log) |
|
|
Describe what is happening with these thermodynamic properties in relation to proteins:
-enthalpy (H) -entropy (S) -Gibbs Free Energy (G) |
-H:
-bond formed, heat produced, negative H -bond broken, heat absorbed, positive H -S: -increase disorder, positive S -decrease disorder, negative S -G: -spontaneous rxn when negative -requires energy when positive |
|
|
Why is the Gibbs' Free E value for CH4 so much higher in water than in an inert solvent?
|
- no bonds except minor VDW forces in inert solvent
- water is more ordered (-S) - water forms H bonds with the CH4 (-H) - causes change in water structure, which changes the heat capacity of the system - water: + 6.3, inert sol: -3.5 |
|
|
If the % residue identity between two similar proteins ________, then the RMSD value tends to __________.
|
-decrease
-increase |
|
|
Why does the heat capacity of water change with relation to hydrophobic molecules?
|
-excess heat is required to "melt" the ice-like water that is ordered around the structure
- water has to separate and organize into a conformation that is not favorable -is proportional to the length of the carbon sidechain |
|
|
What is the estimated energetic value for the Gibbs Free Energy of hydrophobic molecules when they come into contact with water?
|
for ever square angstrom of accessible surface area, it requires 25 cal/mol of E
|
|
|
What is the hydrophobic effect?
|
-those influences that cause nonpolar substances to minimize their contacts with water, and amphipathic molecules, such as soaps and detergents, to form micelles in aqueous solutions
|
|
|
It is enthalpically and entropically __________ for nonpolar items to dissolve in water than in nonpolar media. Why?
|
-favorable
-enthalpy: nonpolar proteins can't H bond to the water, so the water must form an unfavorable "shell" around the proteins to compensate for the H bond loss -entropy: allows water (and the nonpolar protein) to become more disordered |
|
|
The structural analysis of protein/protein interfaces has revealed what?
|
-that the interfaces are often very hydrophobic and exclude water
-VDW forces hold the interfaces together -salt bridges and H bonds are often made across the surface, along with sulfide bonds |
|
|
What did alanine scanning mutagenesis reveal about protein/protein interfaces?
|
1. Make AA mutation, express, purify
2. Binding assay K, convert to GFE 3. Map onto the structure Revealed that a small # of AA at the center of the interface contribute ~85% of the binding energy, so all AA do not contribute equally |
|
|
Describe the "O" ring hypothesis.
|
- residues on the outer edge of the hotspot shield the hotspot from water
- AA residues thus experience a different dielectric constant - Causes polar or charged AA in the hotspot to have huge effects on binding properties |
|
|
The rate of formation of [RL] or Kon, is _____ order while the rate of separation of [RL] to [R] and [L] is ______ order.
|
- second (M-1 s-1)
- first (s-1) |
|
|
The maximum value of the diffusion limited rate constant is ______.
|
- 10^9 M-1 sec-1; which is the upper limit for the speed of association
|
|
|
How can some proteins bind faster than the upper limit of the diffusion limited rate constant?
|
- chaperone assistance
- proteins are not hard spheres and bind in water, not the gas phase - encounter complex software determined that once proteins bump into one another they don't just bounce off but undergo multiple collisions before they diffuse apart |
|
|
Why are Koff rates usually slower than Kon rates?
|
-Koff rates are hindered by the proteins interacting through VDW forces, salt bridges, and H bonds
- Kon rates are mainly hindered by the diffusive properties of the solvent (proteins not bouncing into one another all the time) |
|
|
What is the proposed reaction coordinate for protein-protein interactions?
|
.
|
|
|
What is the formula for the protein equilibrium dissociation constant? What is y when Ka[L] =
1. <<1? 2. >>1? 3. 1? |
1. 0 bound
2. 100% bound 3. 50% bound |
|
|
The maximum value of the diffusion limited rate constant is ______.
|
- 10^9 M-1 sec-1; which is the upper limit for the speed of association
|
|
|
How can some proteins bind faster than the upper limit of the diffusion limited rate constant?
|
- chaperone assistance
- proteins are not hard spheres and bind in water, not the gas phase - encounter complex software determined that once proteins bump into one another they don't just bounce off but undergo multiple collisions before they diffuse apart |
|
|
Why are Koff rates usually slower than Kon rates?
|
-Koff rates are hindered by the proteins interacting through VDW forces, salt bridges, and H bonds
- Kon rates are mainly hindered by the diffusive properties of the solvent (proteins not bouncing into one another all the time) |
|
|
What is the proposed reaction coordinate for protein-protein interactions?
|
.
|
|
|
What is the formula for the protein equilibrium dissociation constant? What is y when Ka[L] =
1. <<1? 2. >>1? 3. 1? |
1. 0 bound
2. 100% bound 3. 50% bound |
|
|
What are some physiochemical perturbants used for protein studies?
|
- pH
- inorganic salts - organic solvents (MeOH, EtOH) - temperature - organic salts, chaotropic agents (guanidine HCl, urea) - MUST be reversible processes! (i.e. you can mess it up but want to get it back together again) |
|
|
When a protein goes from unfolded (denatured) to its native, folded state, what changes occur in thermodynamics with respect to the environment (water) and the protein?
|
- Protein:
- noncovalent bond formation in the protein (-dH) - increase in protein order (- dS) - Water: - water H-bond broken around hydrophobic groups (+dH) - increase in water disorder (+dS) |
|
|
In regards to folding/unfolding, at ambient temperatures for proteins, ____________ is favored and at high temperatures _____________ is favored. Using the Gibbs Free Energy parameters, show the graphical representation for this, as well as how you can determine the unfolding temperature.
|
- ambient= folded
- high T = unfolded |
|
|
What is a good way to measure how a protein unfolds?
|
- use guanidine HCl to denature the protein
- measure the a-helical content using CD - cooperative unfolding happens at a small difference in concentration, and unfolding occurs all at once instead of in steps |
|
|
What does the two-state model for unfolding proteins state?
|
- The "FU" theory
- Proteins are either in the folded or unfolded state and not in between - at any point in the equilibrium unfolding pathway, no more than two states will be significantly populated |
|
|
What type of plot is used to graphically (quantitatively) represent the FU theory?
|
- van't Hoff plot
|
|
|
If you mutate an Ile (which is normally found buried in the native protein strucure) to any of the following AA, what would you expect to happen to the dG of the unfolding reaction (go from F to U)?
- Val - Ala - Asp - Gly |
- Val and Ala would make the dG slightly less (-), or more stable of a reaction, because the R groups are getting smaller and are less hydrophobic than Ile
- Gly has no R group, so it is the least hydrophobic of the uncharged AA, and its dG would be more (-) than Val or Ala - Asp is charged, so even though it is larger, it is OK interacting with water, so dG would be the most(-) |
|
|
In lysozyme studies, what has been proven with mutating AA in the active site of the protein?
|
- that protein residues that contribute to catalysis or ligand binding are not optimized for protein stability
- there is a delicate balance between stability and function |
|
|
What are some common protein folding diseases?
|
- A common feature is a generation of insoluble aggregates (amyloid or amyloid fibrils)
- Transmissible spongiform encephalopathies (TSE) - Creutzfeldt-Jakob - Scrapie - Mad Cow, BSE - Alzheimers - Amyloidoses (deposit AF into various tissues resulting in organ failure or death) |
|
|
In vitro studies regarding amyloid fibrils reveal what about normal protein structure?
|
- beta structure appears to be almost completely sidechain independent and depends on common mainchain interactions
- mutations in proteins that form fibrils are destabilized relative to WT - many proteins can form fibrils in test tubes - cooperative folding may be essential to prevent misfolds and aggregation - protein quality control machinery may break down with age, resulting in more AF diseases |
|
|
What type of plot is used to graphically (quantitatively) represent the FU theory?
|
- van't Hoff plot
|
|
|
If you mutate an Ile (which is normally found buried in the native protein strucure) to any of the following AA, what would you expect to happen to the dG of the unfolding reaction (go from F to U)?
- Val - Ala - Asp - Gly |
- Val and Ala would make the dG slightly less (-), or more stable of a reaction, because the R groups are getting smaller and are less hydrophobic than Ile
- Gly has no R group, so it is the least hydrophobic of the uncharged AA, and its dG would be more (-) than Val or Ala - Asp is charged, so even though it is larger, it is OK interacting with water, so dG would be the most(-) |
|
|
In lysozyme studies, what has been proven with mutating AA in the active site of the protein?
|
- that protein residues that contribute to catalysis or ligand binding are not optimized for protein stability
- there is a delicate balance between stability and function |
|
|
What are some common protein folding diseases?
|
- A common feature is a generation of insoluble aggregates (amyloid or amyloid fibrils)
- Transmissible spongiform encephalopathies (TSE) - Creutzfeldt-Jakob - Scrapie - Mad Cow, BSE - Alzheimers - Amyloidoses (deposit AF into various tissues resulting in organ failure or death) |
|
|
In vitro studies regarding amyloid fibrils reveal what about normal protein structure?
|
- beta structure appears to be almost completely sidechain independent and depends on common mainchain interactions
- mutations in proteins that form fibrils are destabilized relative to WT - many proteins can form fibrils in test tubes - cooperative folding may be essential to prevent misfolds and aggregation - protein quality control machinery may break down with age, resulting in more AF diseases |
|
|
Describe the method of hydrogen-deuterium exchange.
|
- Add D20 (D/H exchange) to protein mix
- Exchanges H for D in the amide backbone of the protein - Quench the reaction by lowering the pH to 2.2 - Quickly detect the increase in mass of the protein via mass spec or the electromagnetic differences via NMR - Theory is that the exchange rate equals the exposure of the particular amide to the solvent (i.e. is the AA buried or exposed?) - Can also determine protein/protein interactions |
|
|
Why do people with renal failure have an increase of beta-2-microglobulin? Why is this bad?
|
- this protein is normally degraded in the kidney
- in people with renal failure, there is a large increase in sera for this protein, which can stimulate deposition in skeletal tissues - this protein (catalyzed by Cu) causes aggregates to form |
|
|
Describe the process which induces beta-2 microglobulin fibril formation. How can this reaction be stopped clinically?
|
- ApoM (normal B2M) binds to copper (Cu) at His-31
- Cu withdraws lone pair e from adjacent Pro-32 N and weakens the amide bond - Allows the Pro-32 to undergo isomerization from cis to trans, forming holo M - holo M converts to M*, which can dimerize with other M* - the M* dimers form fibrils - Can stop this by adding EDTA (chelator) to dialysis patient's regimen to capture Cu |
|
|
The Levinthal paradox states what?
|
- that proteins have a directed path for folding
- if a protein were let go and allowed to form all possible conformations, this would take 10^48 years - know that this doesn't happen because most proteins fold on a range of 0.1 to 1000 sec - Predicts presence of folding intermediates |
|
|
What experiment supported teh Levinthal paradox?
|
- BPTI folding experiment
- First expt. where folding intermediates were captured - irreversible trapping and isolation of normally transient intermediates |
|
|
What is the problem with the FU theory?
|
- classical experiments (CD probing) only averaged the behavior of the protein (thus ended up with either F or U) and didn't resolve atomic detail
- Newer methods (experimental such as H-D exchange and theoretical methods using computer programs) show that U is not one species, but an ensemble or distribution of individual chain conformations |
|
|
Describe folding funnels.
|
- vertical axis states the internal free energy of of a given chain conformation
- ex. sum of H bonds, torsion angle energies, hydrophobic solvation energies - this axis may change depending on the solvent - lateral axes: represent different conformational coordinates - features hills and valleys |
|
|
Using the folding funnel theory, what is the purpose of chaperones?
|
- Chaperones pull the AA chains up energy hills to help the protein get into its native structure
- chaperones increase the yield and formation rate of a protein's native state |
|
|
Many chaperones are what types of enzymes? What is the difference in purpose between small chaperones and larger chaperones?
|
- ATPases
- Small: can delay protein folding before complete biosynthesis and prevent aggregation - Large: prevent aggregation and folding intermediates, also lower kinetic barrier to native state |
|
|
Describe the molten-globule state.
|
- putative folding intermediate
- less compact than native state, but more compact than unfolded state - contains lots of secondary structure - has loose tertiary contacts without tight side-chain packing - bind to ANS |
|
|
What is ANS?
|
- 1-anilino-naphthalene-8-sulphonate
- a hydrophobic probe - binds to a protein which is in the molten globule state |
|
|
When predicting protein structures, sequence alignments only work when what happens?
|
- proteins share at least 20-25% AA identity
|
|
|
What is threading?
|
- When an AA sequence does not share at least 20-25% homology with another protein with known structure, its sequence is threaded through a computer program
- this program contains many structural backbones, and a goodness of fit calculation is done against the sequence and potential structure - a way of determining potential sequence from comparing sidechain interaction with environments |
|
|
What do 3D-1D profile programs do?
|
- determine the following for a given protein structure:
- sidechain environment (potential to be buried, partially buried, or exposed) - subdivide the sidechain based on fraction of polar atoms in the sidechain - secondary structure (a-helix, b-sheet, etc.) - develop a scoring table that determines how well any AA would fit into each position for that profile - align sequence/sequences against the 3D profile |