Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
31 Cards in this Set
- Front
- Back
|
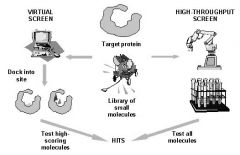
2 Paths to drug discovery
|
|
|
|
In silico drug design
|
computationally "fit" compounds from structural catalog into structure of interest.
-Screen low energy (good fit) compounds, reject high energy compounds |
|
|
Knowing the properties of complexes allows you to calculate or predict:
|
-approximate binding affinity
-binding free energy (deltaG naught, Kd) -binding enthalpy (deltaH naught) -binding entropy (deltaS naught) -kinetic stability (on/off rates) -conformational changes upon binding |
|
|
equation for the free energy of binding:
|
ΔG = ΔH - TΔS
(free energy of binding= enthalpy-(temperature)(entropy) -The more negative ΔG the more favorable the reaction, the tighter the binding. Kd = exp(-ΔG/RT) |
|
|
But we are still a long way from
relating structure and thermodynamics…. |
As thermodynamics is an equilibrium
between free and bound forms and crystal structures are only the bound state… |
|
|
Properties of Drugs binding targets:
|
• Free Energy of Binding is Favorable
• Shape complements binding site • Specific polar and ionic interactions • Still often Entropically driven (hydrophobic, conformationally strained) |
|
|
Things you want to know from the structure of the free enzyme, mutant enzyme with substrate, and WT enzyme with inhibitors
|
free enzyme:
-where is the active site? -how does the ligand bind? -magnitude of induced fit Mutant enzyme with substrates -substrates bind too weakly -do mutations effect structure? WT enzyme with inhibitors -is the inhibitor a suitable lead? |
|
|
Properties we want to predict from structures
|
-drug candidates
-drug targets -ligand-receptor complexes -enzymatic transition states |
|
|
mechanisms we want to understand
|
-enzymatic reactions
-receptor action and modulation -biomolecular interactions |
|
|
Structure Based Drug Design requires:
|
• Need High Resolution Structure (usually Crystal) of Therapeutic Target
• Crystal structure gives the shape of the molecule which may seem trivial but… ~Gives the positioning of the atom types and therefore the flexibility and charge surface of the protein. |
|
|
What does the structure provide (on a basic level)
|
a set of coordinates and atom types from which energy potential can be calculated
|
|
|
3 bonded terms that keep bonds in the correct angles
|
bonds
angles dihedrals -each term used to describe a protein in it's environment |
|
|
dihedral angle
|
the angle between 2 planes
|
|
|
2 non-bonded terms
|
van der Waals
coulombic electrostatics (the only long-range force in nature) |
|
|
applying energetics to drug design:
|
Docking is the goal:
-find the global minimum in the interaction energy between the ligand and the target -accurately estimate the interaction energy -find compounds that bind with the most favorable interaction |
|
|
properties of small molecules you'd like to be able to calculate:
|
-structure and conformations
-solubility (log P) -molecular surfaces -ionization behavior (pKa) -reactivity and stability -toxicity |
|
|
drug-like compounds should follow Lipinksi "rule of 5"
|
<5 H-bond donors
<10 H-bond acceptors <500 Da partition coefficient log P <5 |
|
|
How to perform confomational analysis of small molecule ligands:
|
-find stuctures that are energy minima
-find the global minimum -find structures that are close in energy to the global minimum -estimate relative energies of these structures |
|
|
Analysis of ligand docking
|
-get ligand structures
-fit ligands into target -estimate goodness of fit -estimate binding energy -score ligands based on goodness/energy |
|
|
Docking analysis refinement
|
-keep the protein fixed (bad)
-place the ligand somewhere -evaluate interaction energy or score -find a better ligand position (translate ligand in all directions, rotate ligand, if flexible, change conformation) -stop when cannot improve anymore |
|
|
2 general Steps in drug discovery
|
1) lead discovery
-computationally test compounds from a catalog or custom synthesis -experimentally test good candidates to identify a 'lead' compound 2) lead optimization -alter structure slightly until optimized |
|
|
Vdock technique
|
docking via global minimzation of force-field energy
-define active site with a cube -account for water structure |
|
|
UCSF dock technique
|
1) generate molecular surface of target
2) generate sheres (each sphere is generated tanget to surface point) Cavities mapped by spheres tangential to molecules 3) Select subset of spheres to be used in DOCK |
|
|
How docking works (in silico)
|
-add protons, VDW parameters, and partial charges for both target and small molecule
-create solvent accessible surface area of target -create negative image of surface features surrounding active site using spheres -calculate energy grid for target (each grid point stores VDW score and charge for that area of space) -match ligand atoms to sphere centers and score against grid -rank best scoring poses |
|
|
QSAR
|
Quantitative structure activity relationship
-no 3D structure used, generally a linear equation computed from each molecule, fit to biological activity variations |
|
|
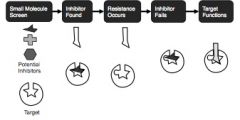
Why Most drugs become obsolete
before their time: |
• Drug Resistance occurs when through rapid evolution a drug target maintains function while no longer being inhibited by the drug.
• Modern Drug Design Fails –> By ignoring function leaves opportunity for drug resistance to occur. • Disrupting the drug targetʼs activity is necessary but not sufficient for developing a robust drug that avoids resistance. |
|
|
Why standard approaches to drug design fail
|
|
|
|
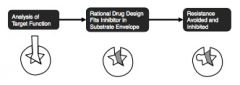
New paradigm of drug design that minimizes chances of resistance
|
|
|
|
HIV-1 protease
|
important drug target as it allows the virus to mature and become infectious
-substrate sites are variable and asymmetric ~multi-drug resistance is common, and occurs where the inhibitors protrude away from the substrate envelope |
|
|
drug resistance
|
a change in molecular recognition
|
|
|
practical steps in designing drugs that are more robust to resistance
|
computationally characterize resistance mutations
-superimpose VDW model surface of substrates -design inhibitors that fit within the VDW substrate envelope |