Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
70 Cards in this Set
- Front
- Back
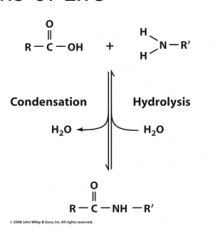
Condensation
|
Elimination of water (amide bond formation, glycoside bond formation)
|
|
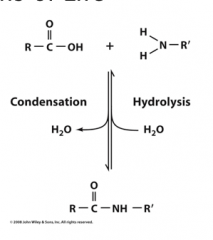
Hydrolysis
|
is cleavage via water
|
|
Self-replication
|
One of the most important features of biological system is that it can replicate itself. Template based synthesis is key for the precise replication of DNA, or for the transmitting of information encoded in DNA to RNA or proteins
|
|
|
Complementarity
|
allows transfer and correction of information.
|
|
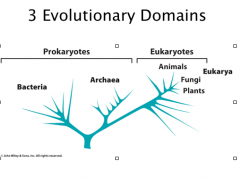
Eukaryotes
Arachaebacteria |
Eukaryotic system: animals, plants and fungal. They are only a very small portion of the organisms on earth
Arachaebacteria (archaea): are groups of bacteria that distantly related to other prokaryotes; often “extremophiles” halobacteria: live in high salt concentration Methanogens: produce CH4 Thermophiles: live in hot springs |
|
|
Evolution
|
Evolution is not directed toward a particular goal = random changes; those that are non-destructive continue
Evolution requires certain degree of flexibility (bio-diversity = less susceptible to one fungal blight) Evolutionary is constrained by its past, which means that evolution is a gradual process Evolution is ongoing. |
|
|
compartmentalization (compartmentation)
|
One of the key features of living organism is that they separate their components from the environments by cell membranes.
Advantages of compartmentation: Separate the cellular environment from the outside Enrich the molecules of interest, for example, nutrients Get rid of toxic molecules by efflux |
|
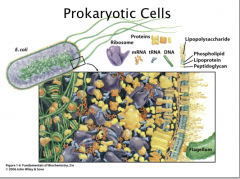
Prokaryotes
|
Prokaryotic cells does not have internal compartments. However, the cytoplasm is not homogenous. The cytoplasm is a very viscous environment so there can still be colocalization of macromolecules and small molecules.
Prokaryotic cells are in general 1 – 10 μm in size. (Human hair = 20-200 um) |
|
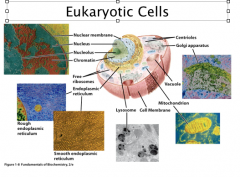
Eukaryotes
|
Eukaryotic cells are 10X bigger, 10 – 100 μm in size.
The key difference between prokaryotic and eukaryotic cells are that the eukaryotic cells have defined nucleus (separated by nuclear membrane). Besides the nucleus, eukaryotic cells can also have the following membrane-separtated compartments: Endoplasmic reticulum: site for the synthesis of many cellular components Golgi apparatus: protein modification and secretion Mitochondria: aerobic metabolism (the power plant in eukaryotic cells) Chloroplast: photosynthesis (plants) Lysosomes: digestion Vacuoles: used for storage (plants, some bacteria) Cytoplasm = cytosol + membrane-bound oraganelles. Organized by cytoskeleton Prokaryotic and eukaryotic cells evolve to suit their living environments. Prokaryotic cell grow fast and can tolerate significant environmental fluctuations; Eukaryotic cells evolved to suit stable environments. |
|

Coupled Reactions
|
Reduction: Gain of elections
Oxidation: Loss of electrons Number of Bonds to O or N −Number of Bonds to H = Oxidation # of Carbon Photosynthesis couples favorable redox reactions to unfavorable reduction of CO2 Catabolism couples favorable redox reactions to unfavorable oxidation to CO2 |
|
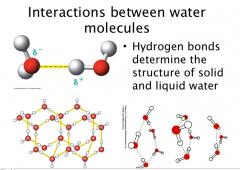
Water
|
Tetrahedral shape if lone pairs are included, or bent.
Polar Hydrogen bonds determine the structure of solid and liquid water (Structured H-bonds in ice Dynamic, moving, switching, temporary H-bonds in water (constantly exchanging)) Hδ+ end of one molecule interactions with Oδ- end of another molecule |
|
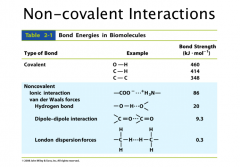
Hydrogen Bonding
|
Van der waals distance is outside edge of electron cloud
Covalent bond is complete overlap H-bond is partial overlap– makes the interaction stronger Need: -a Hydrogen bonded to a very electronegative element (O, F, N) -an electronegative atom with lone pair |
|
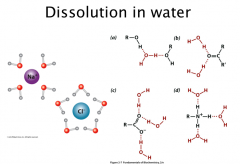
Dissolution in water
|
Ionic and polar substances interact with the dipoles in water: solvation, hydration
|
|

Hydrophobic Effect
|
Spontaneous, Entropy Driven
Water becomes more ordered around non-polar (hydrophobic) = entropy loss ( -ΔS) Hydrophobic species aggregate to minimize surface area contact to water and decrease entropy loss of water. More H2O disorder = + ΔS Spontaneous: ΔH – TΔS = ΔG |
|
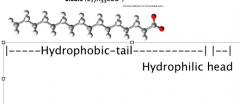
Amphiphiles
|
Amphiphiles are molecules with both polar and nonpolar segments (hydrophilic and hydrophobic)
(Amphipathic) Ex. Lipids (ions of fatty acids—from soaps, w/cation) |
|
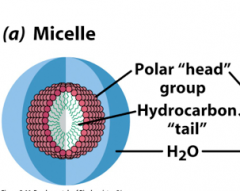
Micelle
|
Ball structure with hydrophobic core and hydrophilic shell
Wedge shaped Lipids tend to form micelles Disrupt these structures with detergents, i.e. sodium dodecylsulfate (SDS)–other amphiphiles –use in lab. Chaotropes: disrupt structure of water: Urea, Guanadinium HCl |
|
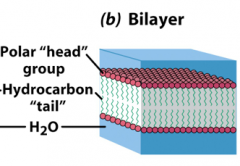
Bilayer
|
Two layers with hydrophobic pore sandwiched in between two layers of hydrophilic segments.
--prevents crossing of polar molecules though hydrophobic layer Cylindrical phospholipids tend to form bilayer Disrupt these structures with detergents, i.e. sodium dodecylsulfate (SDS)–other amphiphiles –use in lab. Chaotropes: disrupt structure of water: Urea, Guanadinium HCl |
|
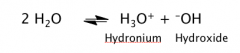
Autoionization of Water
|
H2O dissociates into H+ (H30+) and OH-
H+ can travel long distance by proton jumps (similar to the current move in electron conductors) (move faster than diffusion) That’s why acid/base reaction is the fastest reaction in aqueous solution. |
|
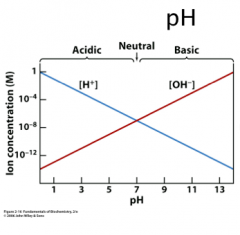
Kw
|
Kw = [H+] [OH-] At 25 0C, Kw = 10-14
So then in pure water at neutral pH the [H+] = [OH-] = 10-7 pH = - log [H+] so pH = 7; acidic pH < 7.0 , basic, pH > 7.0 |
|
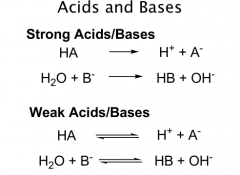
Acids
|
proton donors
Strong acids: transfer all of their protons to H2O Complete dissociation Weak = incomplete dissociation HA = acid A- = conjugate base |
|
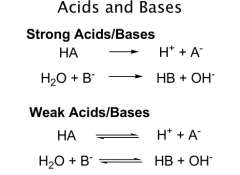
Bases
|
proton acceptors
Strong vs Weak |
|
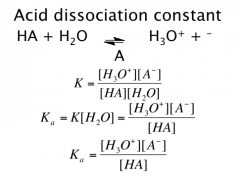
Acid Dissociation Constant
|
Weak acids and bases dissociate only partially
Defined by Ka = tendency to ionize pKa = - log Ka Strong acids have “unmeasurably large” Ka’s *Since strong acids transfer all H+ to H2O, strongest acid in water is H3O+ Strongest base in water is –OH. |
|
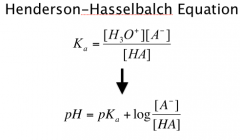
Henderson-Hasselbalch Equation
|
Ratio of acid to its conjugate base
Determine pKa from pH if know ratio |
|
|
pKa trends
|
BELOW pKa, species is mostly protonated
ABOVE pKa, species is mostly Deprotonated AT pKa, 50/50 split (1:1 ratio): |
|
|
Titration
|
http://www.youtube.com/watch?v=tvCyrBHJfBk
|
|
|
Buffers
|
Buffer = Weak acid + conjugate base
1:10 to 10:1 – range in which buffer can most effectively resist changes in pH (addition of acids or bases) Rule of Thumb: Effective buffer when pH = pKa ± 1 |
|
|
Polyprotic Acids
|
Different pKa for each proton removal.
Can only see discrete steps if pKas are sufficiently separated At 1st equivalence point, pH = ½ (pKa1+pKa2) http://www.youtube.com/watch?v=dbddD-qHdTw |
|
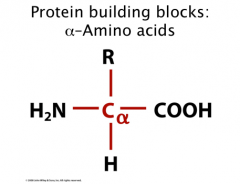
Protein Building Blocks
|
0 “standard” amino acids are building blocks of proteins
Made up of carboxylic acid and amino group on alpha carbon = “alpha-amino acid” “R-group” is side chain specific to each amino acid **Note: Alpha carbon is CHIRAL – has 4 different groups attached Chiral center = tetrahedral with 4 different groups (as long as R is different) |
|
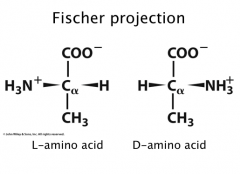
Fischer Projections
|
Rules: carboxyl/carbonyl at top; heteroatoms across from H’s going down (R group at bottom)
Most amino acids are L |
|
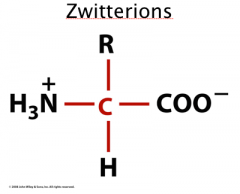
Zwitterions
|
At physiological pH (~7.4) amino and carboxylic acid groups ionize.
pKa (-COOH) = 3.5 pKa (NH3-) = 9.0 Dipolar ions = zwitterion Amphoteric- can act as an acid as well as a base |
|
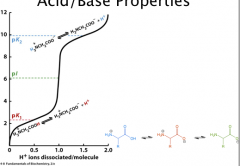
Isoelectric Point
|
pI = the pH at which a molecule carries no net electric charge
With only 2 groups, pI = ½ (pK1 + pK2) Therefore, pI of nonpolar, most uncharged polar is (3.5+9.0)/2 = 6.25 For those with R groups, depends on how charges balance Acidic = ½ (pK1+pKr) Basic = ½ (pKr+pK2) The 2 pKas chosen must be the ionizations that involve the neutral species: If pH is BELOW pI, then protein is PROTONATED (+ charges or neutral = overall positive charge) If pH is ABOVE pI, then protein is DEPROTONATED ( - charges or neutral = overall negative charge) |
|
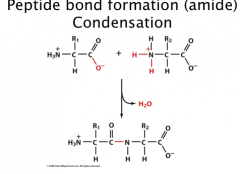
Peptide Bond Formation
|
Condensation reaction (elimination of water)
Di, tri, tetrapeptide Oligopeptide, polypeptide Amino terminus (N-term) Carboxy terminus (C-term) Write N (start) to C (end) |
|
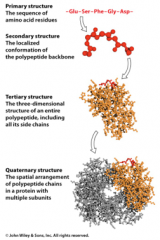
4 levels of protein structure
|
primary, secondary, tertiary, and quaternary
|
|
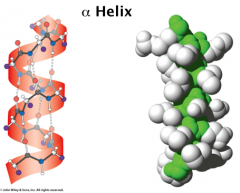
secondary structure
|
Alpha helix:
α helix is right handed, built from adjacent residues Each turn has 3.6 residues and span 5.4 Å on average (rise/pitch) The structure is held together by backbone H-bonds: the C=O bond at n residue H-bonds with the amino group of n+4 residue. α helix is tightly packed and its atoms are in van der waals contact – R groups extend outwards from helix. Pro will kink a helix (helix-breaker) Not Gly—too many other conf (favor b sheet) Steric clashes between large/branched side chains disfavor helix formation (I, Y) With all C=O pointing towards C-term end, helix develops overall charge. N-term is +. C-term is -. http://www.youtube.com/watch?v=Y24qTh4uUHU |
|
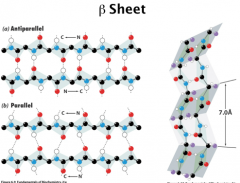
Secondary Structure
|
Beta Sheets
In β Sheet, the H-bonds occur between neighboring peptide chains. Antiparallel β Sheet, the two chain run in opposite direction Parallel β Sheet, the two chain run in the same direction In β Sheet, the side chains of two successive side chains locate on the opposite side of the sheet. β Sheet in proteins can contain between 2 – 22 polypeptide strands with average of 6 strands. Each strand can have up to 15 residues, average 6 residues. Parallel β Sheet rarely has less than 5 strands, which suggests that antiparallel β Sheet is more stable than parallel β Sheet (distorted H bonds) 7.0 A between R groups (pleat) |
|
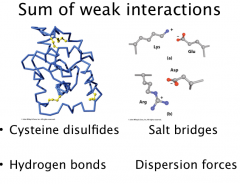
Tertiary Structure
|
3o structure is a collection of 2o structures in motifs or domains
3o str dependent on side chain interactions: Disulfides (covalent) between cysteines (also cross-links between metal ions and ligands Cys, His) Salt bridges (ionic) between acidic and basic residues H-bonds between polar residues Hydrophobic interactions between nonpolar residues Sum of lots of noncovalent interactions hold proteins together in 3D fold. |
|
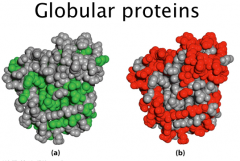
Globular Proteins
|
A hydrophobic core and hydrophilic surface
Hydrophobic effect drives protein folding |
|
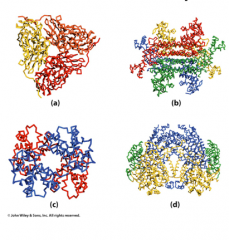
Quaternary structure
|
Multiple subunits held together by primarily non-covalent interactions
Same as tertiary structure interactions |
|
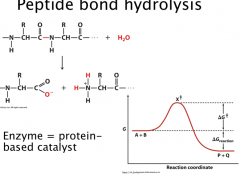
Peptide bond hydrolysis
|
Condensation reaction (elimination of water)
Very slow reaction – thermodynamically favorable, but kinetically controlled Peptide bond has half-life of 20 years Change rate of reaction: increase temperature, increase substrate concentration, add a catalyst Enzyme = protein-based catalyst |
|
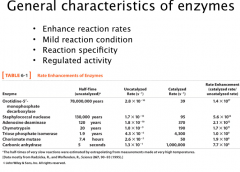
Enzymes
|
General properties of enzymes:
A. Enzyme catalyzed reactions are 10^6 – 10^12 times faster than the uncatalyzed reactions and at least several orders of magnitude higher than chemically catalyzed reactions. B. Mild reaction condition: enzymatic reactions occur at < 100 C, atmospheric pressure, and near neutral pH. Chemical reactions normally need more stringent conditions: high temperature, high pressure and extremes of pH. C. Reaction specificity: for both substrate and product -- distinguish small structure perturbations on substrates. rarely have side products (even stereoisomers) D. Regulation: enzymatic reactions are normally regulated. Common methods used to regulate enzymatic activity: allosteric regulation, post-translational modification of proteins, and up or down regulation of enzyme concentrations. Ex. Carbonic anhydrase CO2 + H2O -->H2CO3 |
|
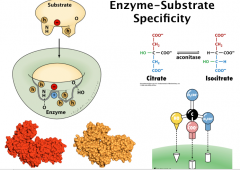
Enzyme-Substrate Specificity
|
Forces used in enzyme/substrate interaction: similar to the forces used in protein folding
A. van der waals interactions B. H-bonding C. electrostatic interaction D. hydrophobic interactions Factors contributing to the specificity in substrate binding: A. geometric complementarity (the shape of the protein binding pocket matches with the shape of the substrate) B. electronic complementarity (negatively charged area on the substrate face the positively charged area in the enzyme) (lock and key) C. induced fit: binding of the substrate can induce conformational changes of the enzyme to accommodate both geometric and electronic complementarity. Geometric specificity more stringent than stereospecificity Enzymes do have some degree of flexibility. For examples, ethanol dehydrogenase can also take methanol or isopropanol as the substrate at a much lower turnover rate. Chymotrypsin can take both peptide and esters as the substrate—however does have geometric specificity aromatic (Y, W, F) just upstream from cleavage site (R-group) Many enzymes permissive—but how permissive is question: Lock-and-key vs. Induced fit “spectrum” |
|
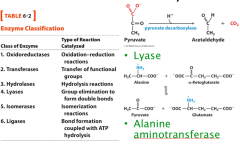
Classifications of Enzymes
|
There are six major classes of enzymes:
oxidoreductase: redox reaction transferase: transferase reaction hydrolase: hydrolysis isomerase: isomerization lyase: group elimination ligase: bond formation at the expense of ATP hydrolysis |
|
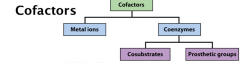
Cofactors
|
Cofactors can be either metal ions or small organic molecules.
Common metal ions in enzymes: Ca2+, Mg2+, Fe3+, Zn2+, Cu2+. The toxic effect of some metals (Cd2+, Hg2+) are due to the replacement of other essential metals by them. Cosubstrate = transient association Prosthetic grp = permanent association Apoenzyme: enzymes after removing the cofactors; holoenzyme: enzyme-cofactor complex. |
|
|
Coenzymes and Vitamins
|
The compounds present in organism’s diet, which are used as the precursors for coenzyme biosynthesis, are called vitamins
Example: Nicotinamide or nicotinic acid are the precursors for NAD+. Supplementation of human diet with nicotinamide relieves the human dietary disease, pellagra (diarrhea, dermatitis, and dementia) All vitamins used to coenzyme synthesis are water soluble vitamins. The water insoluble vitamins are not used in coenzyme biosynthesis. |
|
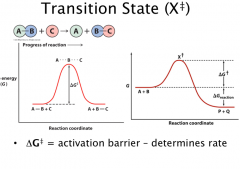
Transition State
|
ΔG‡ and ΔG represent two different concepts in chemistry.
ΔG‡ the free energy of activation. It is the factor governing the reaction rates (Kinetics). ΔG is the free energy of the reaction. It is the factor governing the feasibility of the reaction (Thermodynamics). ΔG < 0, reaction will go to the forward direction ΔG = 0, reaction is at the equilibrium ΔG > 0, reaction will go to the reverse direction |
|
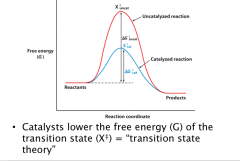
Rate enhancement: ΔΔG‡
|
Transition state theory: for chemical reactions, there exists a state in which a bond is in the process of breaking and another bond is in the process of forming.
ΔΔG≠ decrease by 1.36 kcal/mol (5.71 kJ/mol) will increase rate by 10 fold 10 = e^(5.71/8.3145 J/K mol * 298 K) V = e^(ΔΔG≠ /RT) One important H-bond can increase rate by > 100 fold. (Hbond ~20) 10^6 fold increase with ~34 kJ/mol Small decrease of ΔG≠ can result in dramatic rate acceleration Accelerates both fwd and reverse reactions – allows rxn to reach equilibrium faster |
|
|
Mechanisms of enzyme catalysis
|
Preferential binding of transition state
Proximity effects Acid-base catalysis Covalent catalysis Metal-ion catalysis |
|
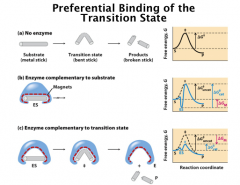
Transition State Theory
|
Transition state theory predicts that enzyme has higher affinity to transition state relative to the ground state or the product.
If a compound mimics the transition state, it should have very high affinity to the enzyme and can be great inhibitors. Based on the transition state theory, the enzyme has preferrential binding to the transition state, which leads to the dramatic decrease of the activation energy. |
|
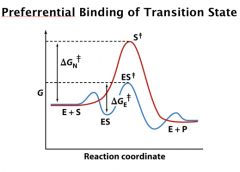
Preferrential Binding of Transition State
|
The most important contribution to the rate acceleration in enzyme catalysis is the preferential binding of enzyme to the transition state.
Stabilizing the transition is the most significant one. Every 1.36 kcal/mol decrease in activation free energy increases the rate by 10 x. Millions of fold increase can be easily achieved. |
|
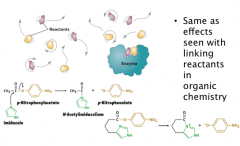
Proximity and Orientation Effects
|
Same as effects seen with linking reactants in organic chemistry
Enzyme can enhance rate by confine the substrate: Bring together the substrate or put the substrate to enzymatic catalytic groups (5 x increase in rate) Proper orientation (e.g. SN2 reaction needs the linear alignment of the reaction groups; slight deviation from that can significant decrease the reaction rate) (5 x increase in rate) Charge groups can stabilize the transition state (electrostatic catalysis); they can also be used for proper orientation. Freeze the translational and rotational motions (107 fold) (Entropic cost—made up with binding energy of ES) In the example, by simply coupling the substrate and the catalyst (imidazole), rate can be increased by 24 fold. |
|
|
Factors that Influence Rate Acceleration in Catalysis
|
In summary, the following factors contribute to rate acceleration in catalysis:
A. proximity (local concentration change after binding) B. orientation of the substrate C. strains generated in binding D. desolvation after binding to the enzyme active site E. Most importantly, the preferential binding of the enzyme to the enzyme active site. |
|
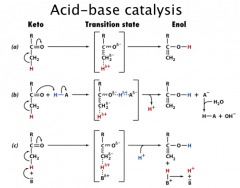
Acid-Base Catalysis
|
General acid catalysis: the process in which proton transfer from an acid lowers the free energy of a reaction’s transition state.
General base catalysis: the process in which the abstraction of a proton by a base lowers the free energy of a reaction’s transition state. Concerted acid/base catalysis: the process in which both general acid and general base are involved |
|
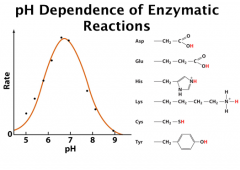
pH Dependence of Enzymatic Reactions
|
This pH dependence curve indicates that the reaction needs both general acid and general base
The left hand side is a general base (dec pH protonate = block needed abstractor) and the right hand side is a general acid (inc pH deprotonate = lose needed H) Ex: Inflection point (pKa) ~ 5.8 and ~ 7.6 Deprot His = gen base Prot His = gen acid pK 4 might be Asp or Glu; pK of 6 might be His; pK of 10 might be Lys. **microenvironments The pH titration curve can also be just half of the above model. |
|
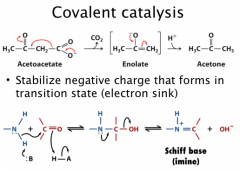
Covalent catalysis
|
Covalent catalysis: it accelerates the reaction rates through the transient formation of a catalyst-substrate covalent bond (covalent substrate)
(nucleophilic catalysis when nuc on catalyst rxts with electrophilic on substrate) In acetoacetate decarboxylation reaction, the amine of a lysine residue function as the base to form Schiff base (Imine) |
|
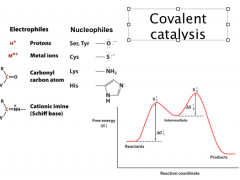
Covalent Catalysis
|
The nucleophilicity is related to the basicity of the functional group (note nucleophiles are deprotonated).
For biological nucleophilic groups, they are either negatively charged or neutral molecules with lone-pair electrons. Good covalent catalysis must balance between nucleophilicity and the ability as a good leaving group, which means that they need to have high polarizability (such as imidazole in His and thiol in Cys) **more stable the bond formed, the harder it is to decompose. Nucleophilicity for starting reaction; good lvg group to release product In addition, coenzymes (e.g., thiamine pyrophosphate and pyridoxal phosphate) function as covalent catalysts. Intermediate (covalent adduct) = different (and lower) path through reaction (ie now two smaller ΔG‡) **Differentiate between intermediate and transition state! |
|
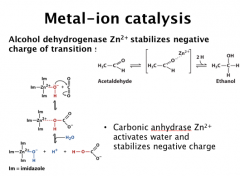
Metal-ion catalysis
|
1/3 of the enzymes are metallo-enzymes. Common metals in enzyme include: Na+, K+, Mg2+, Ca2+, Zn2+, Fe2+/3+, Co2+, Ni2+, Cu2+, Mn2+
Structural roles: Na+, K+, Mg2+, Zn2+, Catalytic roles: Mg2+, Ca2+, Zn2+, Fe2+/3+, Co2+, Ni2+, Cu2+, Mn2+ Roles of metal ions in catalysis: a) orientate the substrate in the enzyme active site; b) mediate oxidation-reduction reactions; c) electrostatically stablizing or shielding negative charges. Example of Zn2+ in H2O activation (metals are normally more potent than proton in neutralizing negative charges. metal ligation to H2O can also significantly increase the H2O acidity) http://www.youtube.com/watch?v=SsI-sQ6Ows8 H2O in carbonic acid anhydrase is activated by Zn2+, in which His64 facilitates this process. This is a very important process in buffer the human body |
|
|
Serine Proteases
|
Serine proteases cleave peptide bonds (hydrolysis)
Catalytic triad: Asp, His, Ser http://www.youtube.com/watch?v=ObivsZ93MNs |
|
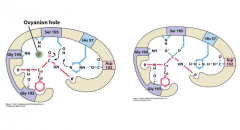
Ex. Serine Proteases
Stabilization of transition state
|
After the formation of the tetrahedron intermediate, peptide move deeper into the active site.
At the same time, there is a new set of H-bonds: the H-bond between oxyanion and the NH of Gly193 and Ser195 backbone. The preferential binding of the transition state contribute significantly to catalysis. Rate enhance: mutants of the triad 5 x 104; native enzyme 1010. |
|
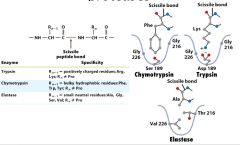
Substrate specificity of serine proteases
|
Chymotrpysin has a big pocket for aromatic residues and Ser189 at the bottom
Trypsin has Asp189 at the bottom to form charge-charge interactions. Elastase: has filled pocket. Thus it take substrates with small side chains. The substrate specificity is not merely controlled by one or two residue. For example, the change of residue Asp189 or Ser189 does not change chymotrypsin to trypsin. Instead, the residues used to form the pocket control the selectivity. (surface loops also replaced to convert trypsin to chymotrypsin-like) |
|
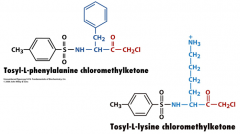
Inhibitors of Specific Serine Proteases
|
By slight modification of the inhibitor, they can be tailed to target specific classes of Ser proteases.
Bulky aromatics for chymotrypsin + chg for trypsin Trypsin, chymotrypsin, elatase = divergent evolution |
|
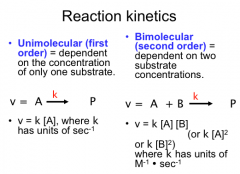
Reaction Kinetics
|
Unimolecular (first order) = dependent on the concentration of only one substrate
Bimolecular (second order) = dependent on two substrate concentrations. |
|
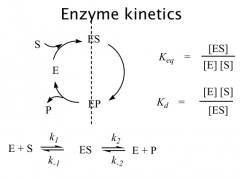
Terms
|
The model of enzymatic reaction can be roughly divided into two sections: binding and catalysis
A few important terms, reaction rate constants (forward and reverse reactions), equilibrium constants and dissociation constants. |
|
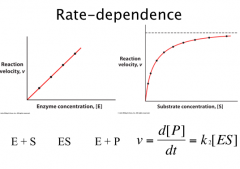
Rate Dependence
|
At high substrate concentration, [ES] = [E]0.
ν0 = k2 [ES] = k2 [E]0. |
|
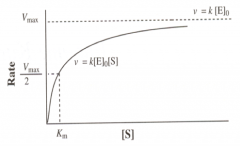
General Features of Enzyme Kinetics
|
At low substrate concentration, it is pseudo 2nd order reaction ν is propotional to both [E]0 and [S].
At high substrate concentration, ν is propotional to [E]0, but independent of [S]. Initial rate and [S] relationship, hyperbolic curve. |
|

|
Definition of the symbols used in enzymatic reactions: S (substrate), P (product), ES (enzyme/substrate complex, Michaelis complex)
The product formation rate depends on [ES] and k2. Without any assumption, this will be a very difficult equation. Under the following initial rate so that the reverse reaction can be ignored A. the rate limiting step is after the substrate binding (k-1 >> k2). B. Rapid equilibrim asumconditions, Michaelis-Menton is derived. C. [S] >> [E], thus, the amount of S present in the form of ES can be ignored. D. Steady state assumption: the concentration of ES remains constant for a long time, which means the formation and decomposition rate are the same (d[ES]/dt = 0). |
|

Michaelis-Menton analysis
|
Assumptions:
k2 is rate-limiting: k2 >>> k-2 and k-1 >>> k2 d[ES]/dt = 0 (Steady state) [E]T = [E] + [ES] |
|
|
Michaelis-Menton Variables
|
Vmax: the maximal velocity of a reaction. It occurs a high substrate concentration and all enzymes are saturated by the substrate.
Km: Michaelis constant and it has the concentration unit. The concentration of the substrate that gives Vmax/2. Km = Kd + k2/k1. Km is very close to Kd when k2 << k-1. The most tricky one is Km. Many people get confused with Kd. In many cases, Km is very close to Kd and used to determine the tightness of the interaction between enzyme and substrate http://www.youtube.com/watch?v=W9o4cQvbShU |
|
![Low-substrate concentration ([S] << Km )](https://images.cram.com/images/upload-flashcards/72/11/69/2721169_m.png)
Low-substrate concentration ([S] << Km )
|
v = k2 [ES]
At low substrate concentrations, the reaction rate has a linear dependence on substrate concentration. |
|
![High-substrate concentration ([S] >> Km )](https://images.cram.com/images/upload-flashcards/72/12/08/2721208_m.png)
High-substrate concentration ([S] >> Km )
|
v = k2 [ES]
At high substrate concentration, the enzymatic rate is substrate concentration independent. |
|
|
variables
|
Km: Michaelis constant and it has the concentration unit. The concentration of the substrate that gives Vmax/2.
Km = Kd + k2/k1. Km is very close to Kd when k2 << k-1. kcat = Vmax/[E]T. Turnover number: this number can change by orders of magnitude. In the above case, kcat = k2. kcat is enzyme concentration independent while Vmax is concentration dependent. |