Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
216 Cards in this Set
- Front
- Back
|
Leigh Syndrome
|
mutations is mitochondrial DNA that causes a degradation in a person's ability to control their movements. Crucial cells in brain stem have mutated mtDNA, creating poorly functioning mitochondria--> chronic lack of energy in cells; usually mutation in OxPhos enzymes or PDH complex, both on X chromosome; treated w/ Vit B1 or thiamin, but infants even with treatment don't usually live past 3
|
|
|
Kearns-Sayre syndrome
|
5,000 bp deletion in mtDNA that is spontaneous (not inherited); causes problems with ETC and OXphos; systemic disorders; eye problems, dysphagia, weakness, hearing loss, etc; no cure, but theurapeutic doses of substances that may mediate electron transfer, such as ubiquinone, vit C and menadione may help
|
|
|
Pearson Syndrome
|
affected mtDNA disorder; causes problems with ETC and OXphos; no cure, but theurapeutic doses of substances that may mediate electron transfer, such as ubiquinone, vit C and menadione may help; muscle weakness & cramping , fatigue, CNS dysfunction, etc are symptoms
|
|
|
Chronic Granulomatous Disease
|
a diverse group of hereditary diseases in which certain cells of the immune system have difficulty forming the reactive oxygen compounds; can't form superoxide and form granulomatas; patients have increased susceptibility to infections; treatment with antibiotics and interferon
|
|
|
Benign Infantile Myopathy
|
affected mtDNA disorder; causes problems with ETC and OXphos; no cure, but theurapeutic doses of substances that may mediate electron transfer, such as ubiquinone, vit C and menadione may help; muscle weakness & cramping , fatigue, CNS dysfunction, etc are symptoms
|
|
|
Complex I: describe
|
Full Name: NADH-CoQ Reductase
Reaction: Reduction, FMN to FFeS, pass E to CoQ 1 at time Point of Entry for: NADH Inhibited By: Amytal, Rotenone H+ translocated: 4 E's translocated: 2 |
|
|
Complex II: describe
|
Full Name: Succinate Dehydrogenase
Reaction: transfers electrons to CoQ Point of Entry for: FADH2 Inhibited By: H+ translocated: 0 E's translocated: 2, to CoQ carrier |
|
|
Complex III: describe
|
Full Name: CytC Reductase
Reaction: CoQ passes E to cytob and c; Reduction Cyto b to Cyto C1 (cytochrome reductase) 2 mol reduced/NADH! Point of Entry for:NADH, FADH Inhibited By: Antimycin H+ translocated:4 E's translocated: 2 |
|
|
Complex IV: describe
|
Full Name:
Reaction:oxidation, reduction, transfer E to O, reduce to Water via Cytochrome Oxidase Point of Entry for:NADH, FADH Inhibited By: CN, CO H+ translocated: 4 E's translocated:2 |
|
|
Complex V: describe
|
Full Name:ATP Synthase
Reaction: synthesis of ATP from protons passing down gradient drives phosphorylation of ADP-->ATP Point of Entry for: NADH, FADH Inhibited By: oligiomyosin |
|
|
ATP Translocase
|
inhibited by atracyloside
|
|
|
AKG and PDH Complex Similarities of Subunits
|
E1: Distinct but similar
E2: Distinct but similar E3: Same! |
|
|
Catalyzes a rxn that results in formation of a high-yield phosphate compound
Choices: A) Malate DH B) Succinyl CoA Synthetase C) Citrate Synthetase D) Pyruvate DH E) Pyruvate Carboxylase |
B) Succinyl CoA Synthetase
|
|
|
Activity is regulated by covalent modifications
Choices: A) Malate DH B) Succinyl CoA Synthetase C) Citrate Synthetase D) Pyruvate DH E) Pyruvate Carboxylase |
D)
|
|
|
Catalyzes an anaplerotic reaction
Choices: A) Malate DH B) Succinyl CoA Synthetase C) Citrate Synthetase D) Pyruvate DH E) Pyruvate Carboxylase |
E) Rxns that increase concentrations of the TCA intermediates without using other TCA intermediates are called anaplerotic rxns; Pyruvate--> OAA is catalyzed by Pyruvate Carboxylase
|
|
|
Catalyzes reduction of nicotinamide adenine to dinucleotide
Choices: A) Malate DH B) Succinyl CoA Synthetase C) Citrate Synthetase D) Pyruvate DH E) Pyruvate Carboxylase |
A) Malate DH catalyzes rxn of Malate to OAA, and NAD+ is reduced to NADH, which rebuilds C4 carbon pool
|
|
|
Which enzymes of TCA cycle reduce NAD+ to NADH?
|
ISODH, AKGDH and Malate DH; NADH can then go on to power OxPHos
|
|
|
Which enzymes of TCA cycle reduce FAD+ to FADH2?
|
Succinate DH; FADH2 can then enter at complex II of OxPhos
|
|
|
Which enzymes of TCA cycle reduce GDP to GTP?
|
Succinate Thiokinase; GTP is converted to GTP--> ATP, which can be used for glycolysis
|
|
|
Define Anaplerotic Reaction:
|
Increase concentration of Citric Acid Cycle Intermediates, which allows for increased rates of oxidation of 2-C units by increasing the overall pool of C-4. As more intermeidates are available, more moles of AcCoA can be processed; an example is Pyruvate Carboxylase, which forms OAA from pyruvate, without depleting other TCA intermediates.
|
|
|
Biotin dependent enzyme of TCA cycle
|
Pyruvate Carboxylase
|
|
|
High serum levels of lactate & alanine suggest?
|
PDH deficiency; they may be formed from lactate DH working in overdrive to compensate for the broke ass OxPhos chain (No working PDH= limited amounts of TCA and OxPhos)
|
|
|
What is ACC? what is it inhibted by?
|
When the enzyme is active, the product, malonyl-CoA is produced and inhibit the transfer of the fatty acyl group from acyl CoA to carnitine with carnitine acyltransferase, which Rroduces malonyl-CoA, which inhibits the beta-oxidation of the fatty acid in mitochondria. Inhibited by the prescense of a fatty acid
|
|
|
pentose phosphate pathway used for?
|
used to provide ribose-5 phosphate for nucleotide biosynthesis OR generates pentose phosphates to enter nonoxidative portion of pathway
|
|
|
GLUT1
|
found in cells with barrier functions; high affinity transporter; KT = 1 mM (serum [glc] = 4-8 mM); GLUT 1 high in RBC
|
|
|
GLUT2
|
in liver, kidney, -cells of pancreas, and serosal side of intestinal mucosal cells; KT = 15-20 mM, functions when [glc]blood is high
|
|
|
GLUT3
|
major transporter in CNS; high affinity
|
|
|
GLUT4
|
in muscle and adipose tissue; insulin-sensitive; KT = 5 mM
|
|
|
GLUT5
|
in small intestine; actually a fructose transporter
|
|
|
General Principles of Glycolysis:
Net ATP? Pi used for? What is formed? What Cofactors are required? |
2 ATP used, 4 ATP produced – Net 2 ATP
Pi taken up and converted to a high energy form by oxidation ATP formed by substrate-level phosphorylation Cofactors (NAD+, ADP) required – must be regenerated for continued operation of pathway |
|
|
What is the Glycerol 3-Phosphate shuttle used for? what organs use it? Yield?
|
Oxidative Regeneration of Cytosolic NAD+
-skeletal muscle, brain Yield = 2 ATP/NADH |
|
|
What is the Malate Shuttle used for? what organs use it? Yield?
|
Oxidative Regeneration of Cytosolic NAD+
-heart, liver, kidney -3 ATP/NADH |
|
|
What is done when O2 is limited?
|
Anaerobic glycolysis;
NAD+ regenerated by lactate DH |
|
|
What are the different tissue distributions of Glycolytic Enzymes?
|
***SKELETAL MUSCLE - high levels of glycolytic enzymes; leads to lactate accumulation when TCA and OxPhos are limiting; low levels of FBPase
***RBCs - glycolysis sole source of energy; lactate (and pyruvate) end products ***LIVER - does not accumulate lactate because of high levels of mitochondria, nutrients and O2; glycolytic or gluconeogenic pathways used; xs glucose--> pyruvate --> fat ; lactate --> glucose **Other tissues - CNS requires glucose --> CO2 + H2O; little or no accumulation of lactate |
|
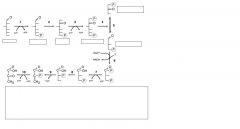
name substrates & enzymes
|
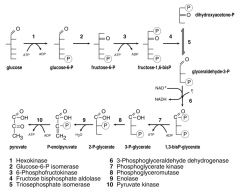
1) Glucose (Hexokinase)
2) Glucose-6-P (Glucose-6-P isomerase) 3) Fructose-6-P (6-Phosphofructokinase) 4) Fructose-1,6,bisP (Fructose bisphosphate aldolase) 5)Dihydroxacetone-P (Triosephosphate isomerase) THEN/OR: 6) Glyceraldehyde-3-P (3-Phosphoglyceraldehyde dehydrogenase) 7) 1,3 bisP-glycerate (Phosphoglycerate Kinase) 8) 3-P-glycerate (Phosphoglyceromutase) 9) 2-P-glycerate (Enolase) 10) P-enolpyruvate (Pyruvate Kinase) PYRUVATE! 11) |
|
|
ovderall yield of ATP in muscle & brain using Gly-3-Phosphate shuttle
|
36 ATP/mol glucose
|
|
|
ovderall yield of ATP in malate shuttle
|
30 ATP/mol glucose
|
|
|
The net effect is purely redox: NADH in the cytosol is oxidized to NAD+, and NAD+ in the matrix is reduced to NADH. The NAD+ in the cytosol can then be reduced again by another round of glycolysis, and the NADH in the matrix can be used to pass electrons to the electron transport chain so that ATP can be synthesized.
|
Net effect of malate shuttle
|
|
|
Enzyme & Regulation:
Glucose --> G6P |
E: Hexokinase
Inhibited by G6P |
|
|
Enzyme & Regulation:
F6P--> FBP |
E:PFK
Enhanced: AMP, F2,6,BP Inhibited: ATP, Citrate |
|
|
Enzyme & Regulation:
GAP<--> 1, 3PGA |
E: 2PGA-DH
Acceptor Control by NAD+ |
|
|
Enzyme & Regulation:
1,3PGA<--> 3-PGA |
E: PGK
Acceptor Control by ADP |
|
|
Enzyme & Regulation:
PEP--> Pyruvate |
E: Pyruvate Kinase
***Acceptor Control by ADP (needed for rxn) ***Allosteric Control by ATP:ADP ratio (high ATP amnt is a negative effector) ****+ Effect by F-1,6-BP |
|
|
explain differences in hexokinase and glucokinase; where are they found?
|
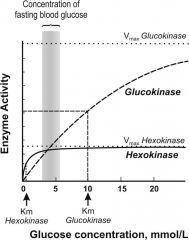
Although both mediate phosphorylation of glucose to G6P, the 1st steps of glycogen synthesis & glycolysis, HEXOKINASE works to convert at much lower glucose [ ] than glucokinase and is much more quickly saturated; hexokinase is found almost everywhere in the body to convert glucose to G6P, but GLUCOKINASE is found in the liver (some in pancreas) b/c its lower affinity for glucose allows liver to better maintain blood sugar levels; glucokinase acts as a glucose sensor, & can trigger shifts in metabolism or cell function in response to rising or falling level sof glucose since it can only phosphorylate glucose if [ ] is high in cell; its Km for glucose is 100x higher than hexokinase
|
|
|
PFK importance: explain. what is it activated by?
|
Phosphofructokinase has historically been regarded as an important control point in the glycolytic pathway, since it is one of the irreversible steps and has key allosteric effectors, AMP and fructose 1,6-bisphosphate (F1,6BP).
--F2,6BP acitvates PFK |
|
|
Explain PFK activation/ inactivation
|
Phosphofructokinase is a kinase enzyme which acts upon Fructose 6-phosphate. There are two types:
1)Phosphofructokinase 1 - converts to fructose-1,6-bisphosphate 2)Phosphofructokinase 2 - converts to fructose-2,6-bisphosphate Fructose 2,6-bisphosphate (F2,6BP) is a very potent activator of phosphofructokinase (PFK-1) that is synthesised when F6P is phosphorylated by a second phosphofructokinase (PFK2). In liver, when blood sugar is low and glucagon elevates cAMP, PFK2 is phosphorylated by protein kinase A. The phosphorylation inactivates PFK2, and another domain on this protein becomes active as fructose 2,6-bisphosphatase, which converts F2,6BP back to F6P. Both glucagon and epinephrine cause high levels of cAMP in the liver. The result of lower levels of liver fructose-2,6-bisphosphate is a decrease in activity of phosphofructokinase and an increase in activity of fructose 1,6-bisphosphatase, so that gluconeogenesis (essentially "glycolysis in reverse") is favored. This is consistent with the role of the liver in such situations, since the response of the liver to these hormones is to release glucose to the blood. |
|
|
What are the roles of Pyruvate Kinase (PK) and Phosphoglycerate Kinase?
|
Pyruvate kinase and phosphoglycerate kinase catalyze the two substrate-level phosphorylation steps, and produce ATP from ADP. While both of these reactions are exergonic, phosphoglycerate kinase is less exergonic (-18.8 kJ/mol) than pyruvate kinase. Phosphoglycerate kinase helps to "pull along" the endergonic glyceraldehyde phosphate dehydrogenase, and in fact, these enzymes are reversible and also function in gluconeogenesis. In contrast, the strongly exergonic pyruvate kinase is irreversible and thus a prime candidate for regulation.
|
|
|
explain basis for anaerobic respiration in humans
|
Under hypoxic conditions(overworked muscles that are starved of oxygen, infarcted heart muscle cells), pyruvate is converted to lactate by anaerobic respiration. This is a solution to maintaining the metabolic flux through glycolysis in response to an anaerobic or severely-hypoxic environment. In many tissues, this is a cellular last resort for energy, and most tissue cannot maintain anaerobic respiration for an extended length of time.
|
|
|
Why is glycolysis alone insufficeint for anaerobic respiration? What must be done?
|
Glycolysis does not regenerate NAD+ from the
NADH + H+ it produces. It is critical for an anaerobic or hypoxic cell to carry out the additional steps of lactate or alcohol production to regenerate NAD+ that is required for glycolysis to proceed. This is important for normal cellular function, as glycolysis is the only source of ATP in anaerobic (RBCs) or severely-hypoxic conditions. |
|
|
The primary functions of the Pentose Phosphate Pathway are:
|
1)To generate reducing equivalents, in the form of NADPH, for reductive biosynthesis reactions within cells.
2)To provide the cell with ribose-5-phosphate (R5P) for the synthesis of the nucleotides and nucleic acids. 3)To metabolize dietary pentose sugars derived from the digestion of nucleic acids as well as to rearrange the carbon skeletons of dietary carbohydrates into glycolytic/gluconeogenic intermediates.(not significant) |
|
|
Where is PPP located?
|
Exclusively in cytoplasm
|
|
|
Explain why RBCs need the PPP
|
The predominant pathways of carbohydrate metabolism in the red blood cell (RBC) are glycolysis, the PPP and 2,3-bisphosphoglycerate (2,3-BPG) metabolism. Glycolysis provides ATP for membrane ion pumps and NADH for re-oxidation of methemoglobin. The PPP supplies the RBC with NADPH to maintain the reduced state of glutathione. The PPP in erythrocytes is essentially the only pathway for these cells to produce NADPH. Any defect in the production of NADPH could, therefore, have profound effects on erythrocyte survival.
|
|
|
The inability to maintain reduced glutathione in RBCs leads to???
|
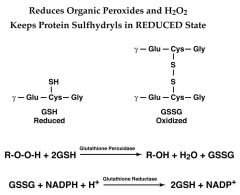
1)Increased accumulation of peroxides, predominantly H2O2, that in turn results in a weakening of the cell membrane and concomitant hemolysis.
2)Accumulation of H2O2 also leads to increased rates of oxidation of hemoglobin to methemoglobin that also weakens the cell wall. Glutathione removes peroxides via the action of glutathione peroxidase. |
|
|
Malaria resistance in African descended people could be due to a deficieny in??? explain why
|
Deficiencies in the level of activity (not function) of glucose-6-phosphate dehydrogenase have been associated with resistance to Malaria; The basis for this resistance is the weakening of the red blood cell membrane (the host cell for the parasite) such that it cannot sustain the parasitic life cycle long enough for productive growth
|
|
|
Name the 5 reacttants, Proudcts and Enzymes of the OXIDATIVE PHASE OF THE PPP
|
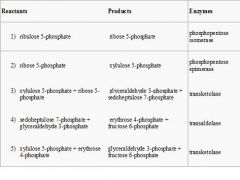
|
|
|
Describe the Oxidative Phase of the PPP pathway (enzymes, reactions, products)
|
Two molecules of NADP+ are reduced to NADPH, utilizing the energy from the conversion of glucose-6-phosphate into ribulose 5-phosphate.
Description of Reactions: 1)Dehydrogenation. The hemiacetal hydroxyl group located on carbon 1 of glucose 6-phosphate is converted into a carbonyl group, generating a lactone, and, in the process, NADPH is generated. 2)Hydrolysis 3)Oxidative decarboxylation. NADP+ is the electron acceptor, generating another molecule of NADPH, a CO2, and ribulose 5-phosphate. 4)Isomerization. (Can also be considered part of nonoxidative phase) Net: Glucose 6-phosphate + 2 NADP+ + H2O → ribulose 5-phosphate + 2 NADPH + 2 H+ + CO2 |
|
|
How is the PPP regulated?
|
***Glucose-6-phosphate dehydrogenase**** is the rate-controlling enzyme of this pathway. It is Allosterically ACTIVATED by NADP+.
***If NADPH needs exceed need for pentose-P, xs pentose-P --> glycolytic intermediates ***If pentose-P needs exceed rate of oxidation rxns, F6P & GAP converted to pentose-P ***If pentoses are in xs in diet, converted to F6P and GAP --Ratio of NADPH:NADP+ ~100:1 in liver cytosol; cytosol is highly-reducing environment. --Formation of NADP+ by a NADPH-utilizing pathway stimulates production of more NADPH. |
|
|
Briefly describe 2 phases of PPP; what is it an alternative to?
|
The PPP is a process that serves to generate NADPH and the synthesis of pentose (5-carbon) sugars. There are two distinct phases in the pathway.
1)Oxidative phase: NADPH is generated 2)Non-Oxidative: synthesis of 5-carbon sugars. PPP is an alternative to glycolysis. It does involve oxidation of glucose, but primary role is anabolic rather than catabolic; takes place in the cytosol |
|
|
What disease does a Glucose 6-Phosphate DH deficieny cause? What are they disease variants? What drugs cause problems and why?
|
--Produces various Hemolytic Anemias as an inherited X-linked disease affecting over 200 million people
*Disease variants: --decreased production of enzyme --decreased catalytic activity --decreased enzyme stability (impt. for RBC) *Oxidative drugs (sulfa drugs, antibiotics, etc.) induce hemolysis in G6PD-deficient cells |
|
|
What disease does a Pyruvate Kinase deficieny cause? explain
|
----Produces Hemolytic Anemias
--ATP levels low in erythrocytes **Patients with high reticulocytes have normal [ATP] Other enzymes of glycolysis are increased High 2,3-BPG levels decrease O2 affinity of hemoglobin |
|
|
What is glucose metabolism important for in the RBC?
|
Glucose metabolism important for:
1)Maintenance of RBC shape 2)Maintenance of Na+/K+ gradients 3)Maintenance of Fe in +2 (ferrous) state 4)Maintenance of reduced sulfhydryl groups 5)Produces 2,3-bisphosphoglycerate (2,3-BPG; a modulator of Hb affinity for O2) |
|
|
What is the Carbon flow of the PPP like? What is the net?
|
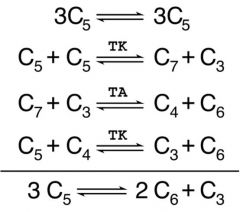
3 ribulose 5-P (3C5) <-->
2 fructose 6-P + GAP 3-P (C3) |
|
|
Phosphorylation steps of Glycolysis?
|
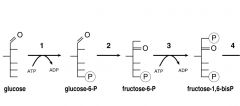
1)Glucose--> 2)G6P--> 3)F6P-->F1,6BP
Enzymes: 1) Hexokinase 2)Glucose-6-P isomerase 3) PFK |
|
|
What are the Cleavage steps of Glycolysis?
|
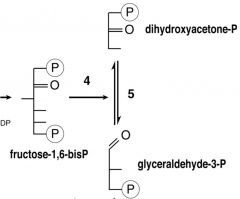
4) F-1,6BP--> DHAP
enzyme: F-BisP-Aldolase 5) DHAP--> GAP enzyme: Trioephosphate isomerase |
|
|
Oxidation Step of Glycolysis?
|
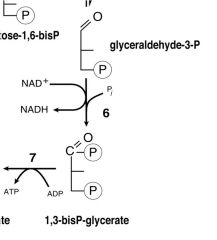
GAP-->1,3BPG
The hydrogen is used to reduce two molecules of NAD+, a hydrogen carrier, to give NADH + H+. |
|
|
ATP formation steps of glycolysis?
|
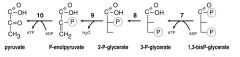
7)1,3BPG--> 3PG
Enzyme: PGK Enzymatic transfer of phosphate group from 1,3-BPG to ADP by PGK, forming ATP and 3PG. At this step, glycolysis has reached the break-even point: 2 molecules of ATP consumed & 2 new molecules synthesized. 1 of 2 substrate-level phosphorylation steps; requires ADP; when cell has plenty of ATP (and little ADP), this reaction does not occur. ATP decays quickly when not metabolized; important regulatory step; Cofactors: Mg2+ 8)3PG--> 2PG E: PGM Phosphoglycerate mutase now forms 2-phosphoglycerate. Enzyme is a mutase; a mutase does not change oxidation state of the carbons. 9)2PG--> PEP E: ENO Cofactors: 2 Mg2+: 1 "conformational" ion to coordinate with the carboxylate group of the substrate, and 1 "catalytic" ion which participates in the dehydration. 10)PEP--> PYRUVATE E: PK A final substrate-level phosphorylation forms 1 pyruvate and 1 ATP by means of the enzyme pyruvate kinase; Additional regulatory step, using covalent modification; Cofactors: Mg2+ |
|
|
steps in PPP that require TPP + Mg++
|
the TRANSKETOLASE reactions that transfer the glycoaldehyde group
happens in non-oxidative steps of PPP, between the isomerase reaction (1st transketolase rxn) and the transaldolase rxn (2nd transketolase rxn) |
|
|
Starch foods:
digested where? by what enzyme? products? |
Where: saliva (1) duodenum (2)
Enzyme: either location uses amylase Products: oligiosaccarides (saliva)maltose (both) |
|
|
Maltose
digested where? by what enzyme? products? |
Where: mucosal epithelium
Enzyme:maltase Products: maltase |
|
|
Sucrose
digested where? by what enzyme? products? |
Where: mucosal epithelium
Enzyme: sucrase Products: glucose, fructose |
|
|
Lactose
digested where? by what enzyme? products? |
Where: mucosal epithelium
Enzyme: lactase Products: glucose, galactose |
|
|
What organ is the major site of unbranched AA degradation & sole site for urea synthesis?
|
Liver
|
|
|
What tissue is the major site for degradation of branched chain AAs?
|
muscle
|
|
|
Which AAs are the major N-containing products that carry N to the liver for conversion to urea?
|
Gln and Ala
|
|
|
DEGRADATION:
Glucogenic AAs can lead to an _____ in __________ Ketogenic AAs can lead to an _____ in _______ |
increase; blood [glucose]
increase; ketone bodies (but NOT glucose!!!) |
|
|
AAs that are degraded to PYRUVATE
glucogenic or ketogenic? |
Ala
Cys Gly Ser Thr *Trp Glucogenic *= not solely |
|
|
AAs that are degraded to:
AcCoA glucogenic or ketogenic? |
*Isoleucine
Leucine *Tryptophan Ketogenic *= not solely |
|
|
AAs that are degraded to:
ACETOACETYL CoA glucogenic or ketogenic? |
Leu
Lys *Phe *Tryp *Tyr Ketogenic *= not solely |
|
|
AAs that are degraded to:
ALPHA-KETO-GLUTARATE glucogenic or ketogenic? |
Arg
Glu Gln His Pro Glucogenic |
|
|
AAs that are degraded to:
SUCCINYL CoA glucogenic or ketogenic? |
*Iso
Met Thr Val Glucogenic *= not solely |
|
|
AAs that are degraded to:
FUMARATE glucogenic or ketogenic? |
Asp
*Phe *Phe Glucogenic *= not solely |
|
|
AAs that are degraded to:
OAA glucogenic or ketogenic? |
Asn
Asp Glucogenic |
|
|
AAs that are degraded to:
PEP glucogenic or ketogenic? |
Serine
Glucogenic |
|
|
AA degradation chart
|
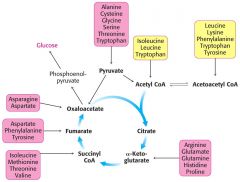
Ser- also PEP
Soley Ketogenic: Leu, Lys Both: 'T TIP' Tyr, Trp, Iso, Phe |
|
|
What is an ENDOPEPTIDASE?
provide specific examples |
A form of PROTEOLYSIS where
SPECIFIC bonds are cleaved --Pepsin – carboxyl side of aromatic and acidic residues --Trypsin – carboxyl side of Lys or Arg --Chymotrypsin – carboxyl side of aromatic and hydrophobic residues |
|
|
What is an Exopeptidase? provide specific examples
|
A form of PROTEOLYSIS where there is NONSPECIFIC cleavage
--Aminopeptidases --Carboxypeptidases --Dipeptidases |
|
|
Maple Syrup Urine Disease
|
Genetic defect – deficiency in branched-chain - ketoacid dehydrogenase
Get build-up of both the branched-chain amino acids and their - ketoacid analogs Appear in urine – give odor of maple syrup Classic form results in neonatal onset encephalopathy |
|
|
Branched Chain AAs
|
Val, Iso, Leu
|
|
|
Write the transamination reaction. What is it used for?
|
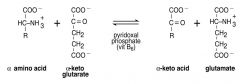
Transaminations transfer an amino group from 1 AA (which is then converted to its corresponding ALPHA-KETO-ACID) to an alpha-keto acid (which can then be converted to its corresponding A-AMINO ACID)
----THUS, N from one AA appears in ANOTHER AA!!!**** this is the beginning step of how the body deals with ammonia, which is a toxic by-product of AA breakdown |
|
|
In transamination reactions, there are ___ pairs of AAs and their corresponding _____; ______ &_______ usually serve as one of the pairs
|
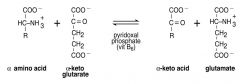
2; A-Keto-Acids; Glutamate and A-Keto-Glutarate
|
|
|
What is the cofactor for transamination reactions?
|
PPP
|
|
|
Explain how nitrogen is removed from glutamate
|
By the glutamate dehydrogenase reaction (other AAs that use transaminations have their own DHs: His, Histidine DH; Ser/Thr, Dehydratase; Asparagine, Asparaginase
|
|
|
How is the Alpha-Keto-Glutarate regenerated in AA metabolism reaction? (draw DEAMINATION rxn, using Glutamate as AA)
|
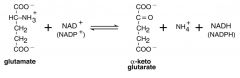
GLUTAMATE DEHYDROGENASE (the enzyme for this rxn) catalyzes the oxidatative deamination of Glutamate , which releases the ammonium ion & forms the A-KG; requires NAD or NADP; other AAs use same process to deaminate; this is a reversible reaction
|
|
|
What controls the rate of Amino Acid breakdown?
|
Regulation of GDH controls rate of amino acid breakdown
--Removal of NH4+ --Acceptor control by NAD+ --Allosteric control: *GTP & ATP inhibit *GDP & ADP reverse inhibition |
|
|
What is the localization of enzymes in the Nitrogen Metabolism pathway?
|
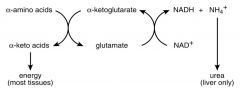
--Aminotransferases: cytoplasm & mitochondria only
--GDH: mitochondrial matrix only |
|
|
What are the sources of NH3? How is it disposed?
|
Sources:
-Diet (bacterial metabolism -Amino Acid degradation in liver - Amino Acid Degradation in other tissues Disposal: -Urea -NH4+ |
|
|
Explain role of Glutamate in Nitrogen removal
|
--Collects nitrogen from other AAs thru transamination reactions
--Nitrogen is released as NH4+, via GDH rxn -- NH4+ & Asp provide N for urea synthesis via the urea cycle |
|
|
Where does Asp get Nitrogen for urea synthesis?
|
Asp gets N from Glu by transamination of OAA
|
|
|
Brief overview of urea synthesis & removal
|
1) Urea formed in urea cycle from NH4+, CO2 & nitrogen of Asp, in the liver
2) NH4+, CO2 & ATP react to form carbamoyl phosphate; enzyme Carbamoyl Phosphate Synthetase I for the rxn is activated by N-acetylglutamate 3) Carbamoyl Phosphate reacts wwith Ornithine to form Citrulline 4) Citrulline reacts with Asp --> Argininosuccinate, releases fumarate & forms Arginine 5) Cleavage of Arginine by arginase releases Urea & regenerates Ornithine |
|
|
Consumming only protein will release:
|
the enzymes of the urea cycle:
--Carbamoyl Phosphate Synthetase I --Ornithine Transcarbamoylase --Argininosuccinate Synthetase --Argininosuccinase --Arginase |
|
|
Draw out the urea cycle; what is the net reaction?
|
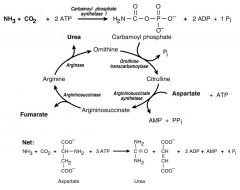
Net:
NH3 + CO2 + Asp + 3 ATP--> Urea + Fumarate + 2 ADP + AMP + 4 Pi |
|
|
What are the fates of fumarate?
|
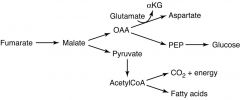
Fumarate can be used to power TCA cycle (Malate--> OAA)
OR to make Fatty Acids (Malate--> Pyr--> AcCoA) |
|
|
What is Ornithine's role in the body?
|
--Reacts with Carbamoyl Phosphate to form Citrulline
--Eventually transported back into mitochondria to be used for next round of urea cycle -- Can be synthesized (if necessary, high protein diet) from Glucose via Glutamate -- Can be used for Arginine Synthesis: Glucose--> Ornithine--> first 4 rxns of Urea Cycle |
|
|
Location of Urea Cycle Reactions???
|
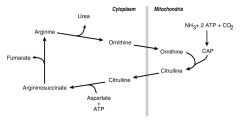
--Carbamoyl Phosphate is stuck in the Mitochondria
--Ornithine & Citrulline can get out of the mitochondria and go to cytosol --Fumarate doesn't go to mitochondria |
|
|
Aminotransferases exist for all amino acids except:
|
Thr, Lys
|
|
|
What is the role of Alanine transaminase?
|
It is important for the delivery of skeletal muscle carbon and nitrogen, in the form of alanine, to the liver. In the liver, Ala Transaminase transfers ammonia to alpha-KG & regenrates PYRUVATE, which can then be diverted into gluconeogenesis
|
|
|
Explain the Glucose-Alanine Cycle
|
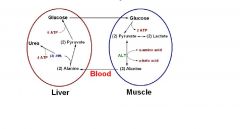
The glucose-alanine cycle is used primarily as a mechanism for skeletal muscle to eliminate nitrogen while replenishing its energy supply. Glucose oxidation produces pyruvate which can undergo transamination to alanine. This reaction is catalyzed by alanine transaminase, ALT. Additionally, during periods of fasting, skeletal muscle protein is degraded for the energy value of the amino acid carbons and alanine is a major amino acid in protein. The alanine then enters the blood stream and is transported to the liver. Within the liver alanine is converted back to pyruvate which is then a source of carbon atoms for gluconeogenesis. The newly formed glucose can then enter the blood for delivery back to the muscle. The amino group transported from the muscle to the liver in the form of alanine is converted to urea in the urea cycle and excreted.
|
|
|
short-term regulation of the urea cycle
|
occurs at CPS-I, which is INACTIVE in the absence of its allosteric activator N-acetylglutamate; steady-state [ ] of N-acetylglutamate set by [ ] pf its components acetyl-CoA & Glu and by Arg, a positive allosteric effector of N-acetylglutamate synthetase
|
|
|
Energy Usage of the Urea Cycle
|
NET TOTAL ATP: UP TO 3 ATP!
--CONSUMES 3 equivalents of ATP and 4 high-energy nucleotide phosphates. --Urea is generated; all other intermediates and reactants are recycled. --Energy consumed is recovered by the release of energy formed during the synthesis of the urea cycle intermediates. --Ammonia released during the glutamate dehydrogenase reaction;coupled to formation of NADH. --When fumarate is converted back to aspartate, the malate dehydrogenase reaction used to convert malate to oxaloacetate generates a mole of NADH; 2 moles of NADH oxidized= 6 moles of ATP. |
|
|
Which reactions of the urea cycle occur in the mitochondria?
|
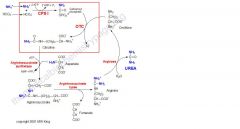
The reactions of the urea cycle which occur in the mitochondrion are contained in the red rectangle. All enzymes are in red, CPS-I is carbamoyl phosphate synthetase-I, OTC is ornithine transcarbamoylase.
|
|
|
What happens to the NH4+ that is transported by Gln?
|
-It is released by Glutaminase where:
KIDNEYS: excrete it as NH4+ in URINE LIVER: conversion to urea SMALL BOWEL: transport to liver for urea synthesis |
|
|
How is Glutamine synthesized?
|
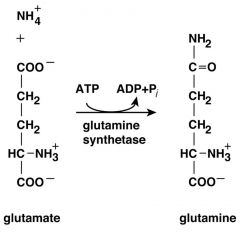
|
|
|
AAs with the highest [ ]s in the Blood?
|
Glutamine: ~8.3
Alanine: ~3.1 |
|
|
Which AAs can go directly to the TCA cycle as a result of catabolism?
|
Go to A-KG
-Glutamine -Glutamate Go to OAA: -Asparagine -Aspartate |
|
|
Which AAs can go directly to the Glycolytic Pathway as a result of catabolism?
|
Alanine
Serine *both to Pyruvate (minor in Ser) |
|
|
Which AAs use Methylmalonyl-CoA as an intermediate of their catabolism before proceeding to TCA cycle?
|
Iso
Val Met Thr |
|
|
brief overview of AA catabolism
-major intermdiates -producuts |
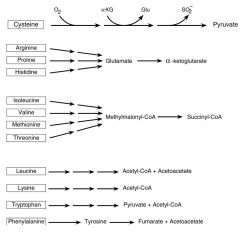
|
|
|
Why does high [glutamate] promotes formation of N-acetylglutamate?
|
because:
Glu + AcCoA N-Ac-Glu + CoASH a positive effector relationship |
|
|
Valine Catabolism
|
1) Partial B-Oxidation
2) 3-Hydroxisobutyrate formation 3) Completion of B-Oxidation 4) Creation of Methylmalonate semialdehyde 5) Conversion to Propionyl-CoA (using CoA-SH) 6)Succinyl-CoA Product |
|
|
Isoleucine Catabolism
|
1) B-Oxidation
2) Beta Cleavage 3) Formation of AcCoA + Propionyl-CoA 4) Succinyl-CoA Product |
|
|
--Explain the role of BIOTIN in AA catabolism
--What is the active form of biotin in this case? |
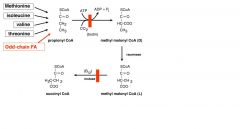
--During catabolism of Val, Iso and Thr, resultant propionyl-CoA is converted to succinyl-CoA for the TCA cycle.
---One of the enzymes in this pathway, methylmalonyl-CoA mutase, requires vitamin B12 as a cofactor in the conversion of methylmalonyl-CoA to succinyl-CoA. --Active form needed is deoxyadenosyl cobalamin |
|
|
What is a Schiff base? Relation to AA catabolism?
|
A Schiff base is a functional group that contains a carbon-nitrogen double bond with the nitrogen atom connected to an aryl or alkyl group—but not hydrogen; formula R1R2C=N-R3, where R3 is an aryl or alkyl group that makes the Schiff base a stable imine.
---PPP is at the active center of a TRANSAMINASE, forms a Schiff Base with the SUBSTRATE in a TRANSAMINASE reaction |
|
|
How does skeletal muscle deal with NH4+ differently than most other tissues?
|
The sequential action of GDH and glutamate-pyruvate Aminotransferase leads to the incorporation of NH4+ into Alanine, which is carried to the LIVER, transdeaminated & converted back to pyruvate (for energy) and NH4+
OTHER TISSUES mostly form Gln (from Gln synthetase) to deal with NH4+, and then take Gln to LIVER, where Glutaminase can hydrolyze Gln back to NH4+ & Glu |
|
|
Metabolites that can't cross the IMM
|
NAD(H), NADP(H), FAD (enzyme bound), all nucleotides (except ATP & ADP), CoA & acyl derviatives, simple sugars, sugar-P, ions (Na+, K+, Cl-) OAA
|
|
|
Metabolites that have SPECIFIC TRANSPORTERS in the IMM
|
1)Pyruvate, TCA cycle intermediates except OAA & succinyl-CoA
2)ATP & ADP via the antiport translocase 3)Aspartate and Glutamate 4)Fatty acids (via carnitine shuttle) 5) Ca+2, Pi 6) Protons – Outward (electron transport) – Inward (ATP formation) |
|
|
Explain how the Mal-Asp shuttle works:
--where --for what?? --enzymes involved?? |
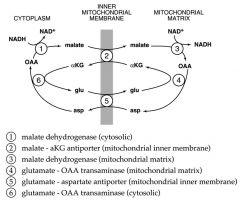
--Each antiport exchanges a C4 for a C5
--No net transfer of either C atoms or N atoms --Only thing transferred in a net fashion is e-’s (reducing equivalents) REACTIONS: a) cytosolic OAA is reduced to malate by NADH, catalyzed by cytosolic malate DH b) Malate enters mitochondria & is re-oxidized to OAA by mitochondrial Malate DH, which generates NADH in the matrix c) OAA CANNOT cross the MM; to return carbon to cytosol, OAA is TRANSAMINATED to Asp, which CAN be transported into the cytosol d) Asp is reconverted to OAA by another transamination reaction e) in the MM, each mole of NADH generates approx 3 moles of ATP via oxidative phosphorylation f) Glycolysis produces 2 moles of NADH per mole of glucose which is equla to ~6 moles of ATP produced by malate-aps shuttle NET: when 1 mole of glucose is oxidized to CO2 and H20, approx 38 moles of ATP are produced via the mal-asp shuttle |
|
|
Explain how the Glycerol-Phosphate shuttle works
--When used --Substrates and enzymes --what tissues --net ATP |
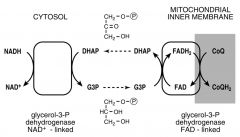
a) Cytosolic DHAP is reduced to Glycerol-3 phoshate by NADH
b) Glycerol-3-Phosphate reacts w/ an FAD-linked DH in the IMM; DHAP is regenerated & re-enters the cytosol c) Each mole of FADH2 produced generates ~2 moles ATP via ox-phos d) Glycolysis produces 2 moles of NADH per glucose, giving a NET of 4 ATP using this shuttle TOTAL: when 1 mole of glucose is oxidized to CO2 and H20, approx 36 mol of ATP is produced using thsi shuttle |
|
|
Coupled Transport Systems
|
--Phosphate (HPO3-) : H+ symport
--Pyruvate : H+ symport |
|
|
Transfer of reducing equivalents between cytoplasm and mitochondria: Name the UNI- & BI- directional shuttles and where they are utlizied
|
--UNIdirectional – Glycerol 3-phosphate shuttle
occurs mostly in skeletal muscle --BIdirectional – Malate-Aspartate shuttle occurs mostly in liver and heart |
|
|
How is energy charge of cell expressed?
|
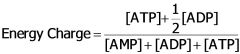
Commonly simplified to [ATP]/[ADP]
|
|
|
The Adenylate system exerts acceptor control in what metabolic pathways?
|
ADP must be available for glycolysis, TCA cycle & OxPhos to proceed
|
|
|
NAD+ exerts acceptor control in what metabolic pathways?
|
NAD+ required for:
glycolysis PDH TCA cycle b-oxidation amino acid deamination |
|
|
FAD exerts acceptor control in what metabolic pathways?
|
TCA cycle
b-oxidation |
|
|
CoA exerts acceptor control in what metabolic pathways?
|
PDH, a-KGDH, branched- chain a-ketoacid DH b-oxidation
|
|
|
What is the purpose Allosteric Controls in metabolic pathways?
|
--Regulation of “Futile Cycles” – coordinated reciprocal regulation
--Reversibility of allosteric control by substrate levels |
|
|
What is regulation by covalent modification? name some examples
|
the addition or removal of a phosphate group by a kinase and phosphorylase, respectively
--either activates or inactivates the enzyme --PDH; actvie in DEphosphorylated state (done by PDH phosphotases) --Pyruvate Kinase (liver) |
|
|
Explain features of MUSCLE METABOLISM at REST:
--Energy charge --affect on regulation points (slowed or increased) --ratio of ATP:ADP --ratio of NADH:NAD and FAD+:FADH2 --overall effect |
HIGH E charge due to low rate of ATP utilization (ATP/ADP INCREASED)
--[ADP] DECREASED - slows glycolysis, TCA cycle, ox-phos. by acceptor control --high ATP/ADP slows following: 1)NADH & FADH2 oxidation 2)PFK 3)PK --isocitrate dehydrogenase - [citrate] INCREASED, which slows PFK ---[G6P] INCREASED inhibiting hexokinase ---high [NADH], [FADH2] slows (acceptor control): glycolysis, PDH complex, TCA cycle, FA oxidation, AA deamination ---Covalent modification slows PDH OVERALL EFFECT: maintain E charge; resting muscle uses FA and KB in preference to glucose |
|
|
Explain features of MUSCLE METABOLISM during VIGOROUS EXERCISE:
--Energy charge --affect on regulation points (slowed or increased) --ratio of ATP:ADP --ratio of NADH:NAD and FAD+:FADH2 --overall effect |
HIGH rate of ATP utilization tends to lower E charge (ATP/ADP DECREASED)
-low ATP/ADP increases/stimulates: 1)Ox-Phos and ETS 2)PFK 3)PK 4)citrate synthase 5)isocitrate dehydrogenase --increased [AcCoA] stimulates pyruvate carboxylase-->C4 pool INCREASED, and TCA cycle INCREASED --[NAD+] & [FAD] INCREASED, stimulating FA oxid’n & formation of more AcCoA --amino acid degradation stimulated by INCREASED [NAD+] |
|
|
What happens when MUSCLES are excersing so vigorously that energy demand is EXCEEDING the maximum rate of electron transport?
|
--E charge is DECREASING, because ATP used faster than it can be regenerated by Ox-phos
--Only glycolysis has excess capacity to generate needed ATP: this is what happens to muscles in this state: 1)NADH accumulates in cytoplasm - regeneration of NAD+ limited by ETS 2)Pyruvate accumulates in cytoplasm - path to TCA limited by ETS 3)NAD+ regenerated in cytoplasm by pyruvate--> lactate 4)Glucose utilization & glycolysis INCREASE 5)FA & AA utilization not increased significantly OVERALL EFFECT: maintains E charge until glucose supply limiting or [lactate] excessive (FA, AA - minor additional E) |
|
|
How are the concentrations of glycolytic enzymes & mitochondria different in red & white muscle fibers?
|
RED:
Glucose is more often converted to PYRUVATE and then enters TCA cycle as OAA or AcCoA (aerobic, oxidative metabolism), has more Pyruvate Carboxylase and PDH ezymes; slow-twitch msucles WHITE: Glucose is more often converted to pyruvate and then LACTATE by LDH; higher [LDH]; fast-twitch muscles |
|
|
Which tissues consume the most energy at rest? at work? constant amount?
|
REST:
--abdominal organs --brain WORK: --skeletal muscle!!!!!! --heart (lesser) CONSTANT: --brain (~.20) |
|
|
What is gluconeogensis? what is it stimulated by?
|
Conversion of metabolic intermediates to glucose by reversal of glycolysis in
gluconeogenic tissues (liver and, to some extent, kidney and intestine) --Helps maintain blood glucose levels --Stimulated by fasting, prolonged exercise, high protein diet, stress |
|
|
Compare energies of Gluconeo & Glyco
|
GLUCONEO:
2 pyruvate-->glucose 2 NADH-->2 NAD+ 6 ATP-->6 ADP + 6 Pi G'= ~-9 kcal/mol GLYCO: glucose-->2 pyruvate 2 NAD+ -->2 NADH 2 ADP + 2 Pi--> 2ATP G'=~-20 kcal/mol |
|
|
Where do rxns of Gluconeo take place in cell?
|
Mitochondrial Matrix:
Pyruvate--> OAA via PC OAA--> Malate via MDH Malate then LEAVES MM and enters the CYTOSOL; CYTOSOL RXNS: Malate--> OAA, via MDH OAA--> PEP, via PEPCK PEP--> DHAP, GA3P--> F1,6,BP -->F6P via F1,6BPase F1,6P--> Glu6P ENDOPLASMIC RETICULUM: G6-P enters ER--> Glucose, via G6Pase GLUCOSE THEN returns to CYTOSOL!!! and goes thru GLUT transporter to enter into BLOOD! |
|
|
How is GLUCONEO generally REGULATED?
|
In General:
Those enzyme-catalyzed reactions that are thermodynamically favorable in one direction only in the cell (i.e. non-equilibrium reactions) are often important sites of regulation |
|
|
how are the reciprocal pathways of gluconeogenesis regulated?
|
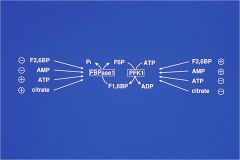
at the F6P<--> F1,6BisP stage
|
|
|
PFK 1 & 2 in fasting state: conditions in cell, what happens to enzyme, what metabolic pathways does this lead to?
|
FASTING STATE:
-PFK2 is phosphorylated by protein kinase A, which is activated by cAMP -Phosphorylated PFK2 converts F2,6 BisPhos--> F6,P which causes levels of F2,6BisPhos to FALL -Lower levels of F2,6BisPhos make PFK1 LESS ACTIVE --PFK2 acts as a PHOSPHATASE in the fasting state, when it is phosphorylated --Gluconeogenesis is STIMULATED |
|
|
PFK 1 & 2 in fed state:
conditions in cell, what happens to enzyme, what metabolic pathways does this lead to? |
--Insulin causes phosphotases to be stimulated
--Phosphotases DEphosphorylate PFK2, which causes MORE conversion of F6P to F2,6BP --F2,6BP levels rise in cell, which makes PFK1 MORE active --Thus, PFK2 acts as a KINASE in the FED state, when it is dephosphorylated |
|
|
PFK1 is activated by:
|
Fructose 2,6 BP.
|
|
|
what happens after eating a meal?
|
--After eating, F2,6BP is formed from F6,P via action of PF2.
--F2,6BP activates PFK1 --PFK1 converts Frucotse-2,6-BP to Fructose-1-6-P --Glycolysis is stimulated |
|
|
PFK1 is activated by AMP and inhibited by ATP; how is this important to muscle?
|
It is a regulatory mechanism;
-During exercise, AMP levels are high and ATP levels are low -Since PFK1 is stimulated by high AMP levels, PFK1 will increase its reaction rates -this increase in activty promotes GLYCOLYSIS which will generate more ATP for the exercising muscle --When ATP is high, high rates of glycolysis aren't needed |
|
|
How is gluconeogenesis/glycolysis regulated at the PEP<--> Pyruvate Stage?
|
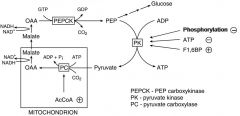
PK
PC |
|
|
Insulin INCREASES
|
glucose uptake (e.g. muscle and fat cells)
glycogen synthesis in liver and muscle amino acid uptake and protein synthesis in most tissues triglyceride synthesis in liver and fat cells |
|
|
Insulin DECREASES
|
gluconeogenesis in liver
glycogen breakdown in liver and muscle triglyceride breakdown in fat cells |
|
|
Glucagon DECREASES (in liver)
|
Glycogen synthesis
Glycolysis Fatty acid synthesis |
|
|
Glucagon INCREASES (in liver)
|
Glycogen breakdown
Gluconeogenesis b-oxidation |
|
|
EPINEPHRINE:
Produced by: Secretion stimulated by : Actions include: Stimulates: |
Produced by: adrenal medulla
Secretion stimulated by : --Stress --Trauma --Extreme exercise --Low blood glucose Actions include: --Increasing blood glucose Stimulates: --Glycogen breakdown in liver and muscle --Triglyceride breakdown in fat cells --Gluconeogenesis in liver --Fatty acid b-oxidation in liver and muscle |
|
|
GLUCCORTICOIDS: (Cortisol)
Produced by? Stimulated by? Actions? Stimulates? |
Produced by adrenal cortex
Secretion stimulated by : Stress Actions include: Changes in gene expression Increasing blood glucose Stimulates: Gluconeogenesis in liver Protein breakdown |
|
|
Give summary of cell surface receptors
|
--Includes insulin, glucagon, epinephrine and lipolytic hormones
--Cannot transverse cell membranes --Bind to cell surface receptors --Leads to generation of “second messengers” (intracellular signals, e.g. cAMP, Ca2+) --Allows amplification of the original signal |
|
|
Give summary of how HIGH blood glucose affects the body
|
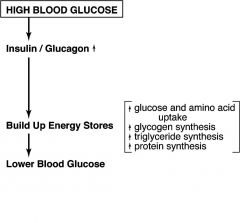
|
|
|
Give summary of how LOW blood glucose affects the body
|
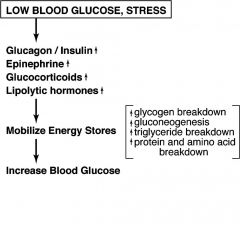
|
|
|
Hormones that stimulate Adenylate Cyclase:
LIVER: MUSCLE: ADIPOSE: |
LIVER:
*Glucagon + *Epinephrine +/- MUSCLE: *Epinephrine + Glucagon - ADIPOSE: Lipolytic Hormone + *Epinephrine + *Glucagon - |
|
|
What are Phosphodiesterases?
What do they do to the cell? |
--Catalyze the hydrolysis of cAMP
-----cAMP + H2O AMP --Allows the cell to return to a basal state --Inhibition keeps the cell in the “excited state” (e.g. caffeine) |
|
|
What is Protein Kinase A (PKA)?
|
Protein Kinase A (aka cAMP-dependent protein kinase)
--Allosterically activated by cAMP --phosphorylates Ser & Thr on several proteins in metabolism --Both Pyruvate Kinase & Bifunctional enzyme are PKA targets in the liver |
|
|
What is the role of Ca2+ in metabolism?
Intracellular levels? Increased by? Stimulates? Action? |
Intracellular levels: Epinephrine (liver)
increased by: Contraction (skeletal muscle) Stimulates: Glycogen breakdown Mode of action: Allosteric activation of phosphorylase kinase |
|
|
How do seteroids enter cells?
Explain and give examples |
Steroids (e.g. cortisol) easily enter cells and bind to intracellular receptors
Hormone-receptor complexes act primarily by altering expression of target genes These include metabolic enzymes, such as: a.Transaminases in skeletal muscle (facilitate mobilization of amino acids) b.PEP-carboxykinase in liver (facilitates gluconeogenesis) |
|
|
How is the insulin signal spread thruout the body?
|
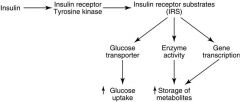
-Membrane receptor has tyrosine kinase activity
-Leads to phosphorylation of multiple substrates -Promotes energy storage -Suppresses gluconeogenesis (e.g. antagonizes glucagon effects) |
|
|
Compare Gluconeogenesis/Glycolysis pathways
|
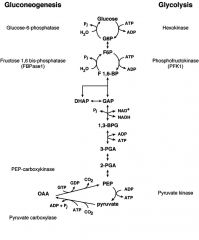
|
|
|
Andersen's Disease (IV)
|
Defect in branching enzyme (liver, spleen)
Normal glycogen amount, but with very long outer branches Progressive liver cirrhosis, early death |
|
|
Cori's Disease (III)
|
Defect in debranching enzyme (muscle, liver)
Abnormal glycogen structure (short outer branches), increased amount Enlarged liver (hepatomegaly), heart (cardiomegaly) Mild hypoglycemia due to inefficient glycogen breakdown Growth retardation |
|
|
Her's Disease (VI)
|
Defect in glycogen phosphorylase (liver)
Increased amount of glycogen Enlarged liver (hepatomegaly) Mild hypoglycemia due to decreased glycogen breakdown |
|
|
McArdle's Disease (V)
|
Defect in glycogen phosphorylase (muscle)
Moderate increased amount of muscle glycogen Normal fasting blood glucose levels Exercise induces painful cramps, release of enzymes indicate muscle damage Low exercise-induced lactate release |
|
|
Von Gierke's Disease (I)
What other types of defects have similar phenotypes? |
Defect in glucose 6-phosphatase
--Increased amount of liver glycogen (normal structure) --Severe fasting hypoglycemia --Most common glycogen storage disease (1 in 200,000) |
|
|
Pompe's Disease
|
Defect in lysosomal a-1,4-glucosidase
Increased amount of glycogen in almost every tissue (normal structure) Normal blood glucose levels Severe cardiomegaly, early death Purpose of this lysosomal pathway of glycogen degradation is not known |
|
|
Defects that give similar phenotype to Von Gierke's Disease?
|
--Defects in carriers transporting G6P, glucose, or Pi across the ER
membrane have similar phenotypes |
|
|
Diseases that affect PHOSPHORYLASE DEBRANCHING ENZYME:
|
-McArdle's: Muscle
-Her's: Liver - Cori's |
|
|
diseases that affect Glycogen synthase branching enzyme
|
Andersen's
|
|
|
Diseases that affect G-6-Phosphate (of liver)
|
von Gierke's
|
|
|
Glucosyl units are linked by?
Branching enzymes form what? |
a-1,4-glycosidic bonds & a-1,6-glycosidic bonds: these are formed by the branching enzyme
|
|
|
What is the purpsoe of branching in glycogen?
|
It increases solubility & # of sites for synthesis & degradation
|
|
|
Basic steps for glycogen synthesis (glycogenesis)
|
1) Glucose--> Glu-6-P via Glucokinase (Liver) or Hexokinase and ATP
2) Glu-6-P--> Glu-1-P via Phosphoglucomutase & ATP 3) Glu-1P--> UDP-Glucose via Glu-1-P-UDPase, UTP and PPi 4) UDP-Glucose + Glycogen (n residues)--> Glycogen + UDP (n +1 residues) via Glycogen Synthase 5) UDP + ATP--> UTP + ADP via Nucleoside Diphosphokinase OVERALL: Glucose + 2ATP + Glycogen(n res)--> Glycogen(n+1 res)+ 2ADP +2Pi |
|
|
Basic Pathway for Glycogen BREAKDOWN:
|
1) Glycogen Phosphorylase [requires vitamin B6 (pyridoxal phosphate)]
Catalyzes the phosphorolysis of a-1,4 glycosidic bonds 2) Glycogen (n residues) + Pi--> Glycogen (n-1 residues) + Glucose 1-P |
|
|
How does debranching take place?
|
1)an a-1,6 Glycosidic Bond gets broken where it is linked to core via a PHOSPHORYLASE, which leaves Glu-1-P. It takes 8 Pi to do this
2) Transferase transfers the a-1,4-linked branch to core 3) a-1,6 branch broken using a-1,6 Glucosidase |
|
|
Glucose 6-P is used differently in the body. explain
|
Glucose 6-P makes Glucose for the liver (to regulate blood glucose levels) but for skeletal muscle, product goes towards glycolysis (to create energy)
|
|
|
Why is Glycogen Phosphorylase NOT regulated by SUBSTRATE AVAILABILITY?
|
Glycogen(n)+ Pi-->
Glycogen(n+1) + Glucose 1-P via GLYCOGEN PHOSPHORYLASE --Glycogen phosphorylase is associated with glycogen particle --Glycogen phosphorylase is normally saturated with glycogen --intracellular Pi is really high already & GlyP is saturated normally |
|
|
Similarities & Differences between Liver & Muscle Glycogen Phosphorylase
|
Differences:
-Encoded by different genes -Liver enzyme not regulated by AMP, glucose 6-P or ATP -Phosphorylation of liver enzyme stimulated by glucagon -Liver phosphorylase a is allosterically inhibited by glucose Similarities: Phosphorylation (b a) is stimulated by epinephrine a form is more active |
|
|
How is Glycogen Phosphorylase activity modified covalently?
|
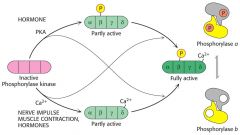
|
|
|
How is UDP-glucose a regulator of glycogen synthesis?
|
It is need for the reaction:
Glycogen(n) + UDP-glucose --> Glycogen(n+1) + UDP --Inracellular UDP-glucose levels are very low normally, but INCREASE with increasing glucose availability --Therefore, GLYCOGEN SYNTHASE activity is dependent on intracellular glucose availability |
|
|
What are three signals for protein degradation?
|
1. Amino terminal amino acid: proteins with certain amino termini turn over
rapidly 2. Proteins with PEST sequencesL: proteins w/ lots of proline, glutamate, serine and threonine have short intracellular half-lives. 3. Denaturation, oxidation or covalent modification (by ubiquitin) are signals for preferential degradation. |
|
|
Ubiquitin's role in cells
|
degrading protein as apart of a proteasome complex, by cross-linking to AA acid groups w/ lysine side chains
|
|
|
a)Lysomes degrade proteins by:
b)Proteasomes degrade proteins by: |
a)a mixture of proteases (cathepsins)
b) releasing peptides & ubiquitin |
|
|
Proteasomes consist of
|
regulatory & catalytic particles, each of multiple subuntis; enzymatic core lies in the center of the cataltic unit;
|
|
|
What are some selectivity features of the ubiquitin pathway?
|
1. E2 carrier proteins and E3 enzymes are specific for different types of proteins.
2. E3 is the “reader” of destabilizing signals such as the nature of the N-terminus. 3. The complex subunit structure of the regulatory particle recognizes ubiquitin-conjugated proteins. 4. Only unfolded proteins can enter the proteasome active site in the catalytic particle. |
|
|
What are the roles of protein turnover in biological processes?
|
A. Quality Control
B. Cell Cycle Regulation C. Inflammation and the Immune Response D. Adaptation to New Physiologic Conditions E. Oncogenesis |
|
|
How is ubiquitin a key player in the inflammation and immune response?
|
1. I-κB, a transcriptional inhibitor, is degraded by proteasomes in response to
inflammatory signals. 2. Peptides resulting from protein (virus) breakdown can reach the cell surface bound to the major MHC1 complex and be presented to cytotoxic lymphocytes. |
|
|
How is ubiquitin a key player in adaptation to new physioligic conditions?
|
1. In periods of fasting, gluconeogenic enzymes are induced, but these are
rapidly degraded upon feeding. 2. Tissue morphogenesis and remodeling during development. Apoptosis inhibitors are selectively degraded. |
|
|
How is ubiquitin a key player in oncogenesis?
|
At least one oncogenic virus targets a tumor suppressor (p53) for destruction, by
the ubiquitin pathway. Mutations that prevent proteasomal degradation of an oncoprotein (Jun) lead to tumors. |
|
|
How is ubiquitin a key player in quality control?
|
Selective degradation of abnormal proteins(hemoglobinopathies, ion channel in cystic fibrosis).
|
|
|
How is ubiquitin a key player in cell cycle regulation?
|
Programmed destruction of regulatory proteins, including cyclins, regulates critical
points in the progression of cells through the mitotic cycle. |
|
|
What are the general tactics for amino acid catabolism?
|
1) Transamination (ex alanine)
Alanine + α-Ketoglutarate ⇔ Pyruvate + Glutamate 2) Conversion to intermediate, followed by transamination (ex asparagine) Asparagine + H2O ⇒ Aspartate + NH4+ Aspartate + α-Ketoglutarate ⇔ Oxaloacetate + Glutamate 3) Oxidative deamination of glutamate (i.e. glutamate dehydrogenase) Glutamate + NAD(P)+ ⇒ α-Ketoglutarate + NAD(P)H+ + NH4+ 4) Conversion to glutamate, followed by oxidative deamination (e.g.glutamine) Glutamine + H2O ⇒ Glutamate + NH4+ Glutamate + NAD(P)+ ⇒ α-Ketoglutarate + NAD(P)H+ + NH4+ 5) Direct deamination of the amino acid, or anintermediate (e.g.threonine) |
|
|
What is the fate of AAs under conditions of energy excess?
|
1. Resynthesis into protein and synthesis of other body constituents.
2 Conversion to ketoacids and replenishment of TCA cycle intermediates. 3. Conversion of carbon skeletons to glycogen (indirect pathway of glycogenesis). 4. Conversion of carbon skeletons to fatty acids. |
|
|
What is the fate of ketoacids under conditions of energy needs (low blood glucose)?
|
1. Conversion of carbon skeletons to TCA cycle intermediates.
2. Major source of the carbons for net glucose synthesis in LIVER. |
|
|
What is the fate of AAs resulting from protein breakdown in the FASTING state?
|
1. Muscle preferentially uses branched-chain amino acids (leucine, isoleucine, valine) for energy and exports alanine, glutamine and
glutamate to liver. 2. Kidney metabolizes glutamine to help maintain acid-base balance. 3. Liver produces urea from amino acid degradation and glucose by gluconeogenesis (glucagon and glucocorticoids induce some enzymes needed for gluconeogenesis). |
|
|
What is the fate of AAs resulting from protein breakdown in the muscle-wasting catabolic states (sepsis, burns, cancer, etc?)
|
1. Preferential loss of protein from skeletal muscle and skin
2. Predominantly long-lived actin and myosin degradation via the ATPdependent proteasome pathway. Muscle ubiquitin concentrations increase. 3. Glucocorticoids are required for the increase in ubiquitin and proteasome subunit mRNAs |
|
|
Diabetes promotes signals for?
|
store mobilization, incl. increased protein breakdown
|
|
|
How does cortisol regulate protein turnover?
|
Increases during catabolic states
Promotes protein breakdown, amino acid catabolism, and gluconeogenesis through induction of: a) Ubiquitin b) Proteasome complex components c) Transaminases d) PEP carboxykinase e) Fructose 1,6-bisphosphatase |
|
|
How does insulin regulate protein turnover?
|
--Increases during anabolic states
--Promotes protein synthesis and inhibits protein breakdown |
|
|
How does starvation regulate protein turnover?
|
1. Increased need for blood glucose increases protein breakdown initially.
2. Increased cortisol increases the concentration of enzymes involved in protein degradation and gluconeogenesis (ubiquitin, proteasome complex components, transaminases, PEP carboxykinase, fructose-1,6-bisphosphatase). |
|
|
How does diabetes regulate protein turnover?
|
Absence of or resistance to insulin promotes signals for store mobilization, including
increased protein breakdown. |
|
|
What is the most abundant AA in circulation? What are its main roles?
|
Glutamine
1. Utilized by rapidly dividing cells (e.g. intestinal enterocytes, lymphocytes) Source of nitrogen (nucleotide biosynthesis) and carbon (energy) 2. Carrier of nitrogen e.g. skeletal muscle to liver during starvation 3. Aid in acid/base balance (kidney) 4. gluconeogenic precursor |
|
|
How does "fat burn in the flame of carbohydrate?"
|
In LIVER, the use of C4
acids for gluconeogenesis slows the TCA cycle. Acetyl-CoA builds up as a result of fatty acid metabolism and the metabolism of some amino acids (ketogenic). However, the CoA pool is limited. If free CoA is not available for the carnitine acyltransferase II and ketothiolase reactions, then fatty acid oxidation, and subsequently gluconeogenesis, would stop. The formation of ketone bodies from acetyl-CoA frees CoA for continued β-oxidation |
|
|
Where does gluconeogenesis occur? What is it in response to?
|
Liver;
METABOLIC SIGNALS to release glucose into blood; requires a carbon source (C4 acids) and HIGH energy charge (lots of ADP)C4 acids replenished by glucogenic amino acid breakdown. ATP and NADH are required to drive gluconeo-genesis. Both generated from β-oxidation of fatty acids (and subsequent oxidative phosphorylation for ATP generation). Acetyl-CoA generated from β-oxidation promotes gluconeogenesis by allosterically activating PYRUVATE CARBOXYLASE. |
|
|
STARVATION CONDITIONS:
REQUIRE? TACTIC? HORMONAL RESPONSE? METABOLIC RESPONSE? |
Require:Maintenance of blood glucose levels
Tactic: --Decreasing peripheral tissue glucose utilization --Increasing hepatic glucose production Hormonal: --Decreased insulin --Increased glucagon --Increased epinephrine and lipolytic hormones --Increased cortisol Metabolic: --Increased lipolysis and ketogenesis supply alternative fuels --Increased hepatic b-oxidation drives gluconeogenesis --Increased protein breakdown |
|
|
draw out starvation conditions in FAT, LIVER and MUSCLE
|
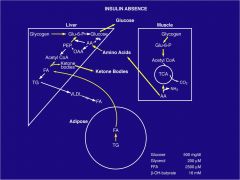
|
|
|
draw out the inflammtion response that activates ubiquitination
|
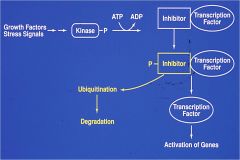
|
|
|
draw out path to energy generation in response to high cell charge
|
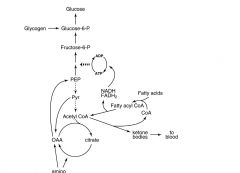
|