Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
77 Cards in this Set
- Front
- Back
|
Renal cell carcinoma
-presentation -3 types |
•Classic triad (less than 10%)
–Hematuria –Flank mass –Flank pain •Other symptoms –Anemia (normochromic/normocytic) –Fever –Weight loss –Polycythemia –Hypercalcemia –Hepatic dysfunction unrelated to presence of metastatic disease (“Stauffer syndrome”) •Histological subtypes of renal cell carcinoma: –Clear cell (70-80%) –Papillary (10-15%) –Chromophobe (5%) •Risk factors –Cigarette smoking –Obesity –Dialysis –Genetic (von Hippel Lindau, others) RX- surgical resection for stage III, immunologic therapies for stage IV (IL-2) |
|
|
IFN-a 2B
-indications -SE -Pegylated |
Immune therapy for melanoma and renal carcinoma
STAGE 3 MELANOMA •Used for adjuvant therapy of Stage III melanoma and therapy of Stage IV renal cell carcinoma •Shown to have improvement in relapse-free survival in melanoma •Benefit on overall survival in melanoma is less clear •Mechanism of action unclear •Development of autoimmune effects (vitiligo or development of autoantibodies) associated with response •Possible antiangiogenic effects? •First month: 20 million IU Daily IV infusion 5 days/week for 4 weeks •Then 10 million IU SQ injection 3 days/week for 48 weeks •Data suggest that the first month provides the greatest benefit SE •Constitutional: fatigue, fever, anorexia, weight loss, and malaise. •Myelosuppression –Neutropenia associated with interferon does not necessarily correspond to increased rates of infection. •Hepatotoxicity •Thyroid dysfunction •Neuropsychiatric effects Pegylated IFN •Associated with fewer adverse effects •Improved relapse-free survival •Has not shown improvement in overall survival •Weekly dosing for up to 5 years |
|
|
IL-2 in cancer treatment
-indications -SE |
Immune therapy for melanoma and renal carcinoma
•Used in Stage IV melanoma and Stage IV renal cell carcinoma with CLEAR CELL HISTOLOGY •RR: 16% (6% CR with duration 2-5 yrs) •Limited applicability •Healthy patients •Specialized administration centers •M1a and M1b disease most responsive •Mechanism poorly understood –Immunostimulatory effects –Direct anti-tumor effects •Administered as an inpatient for close monitoring and careful assessment prior to each dose. •A cycle consists of 14 planned doses given 8 hours apart on days 1-5 and 15-19 •Most patients end up receiving 7-10 doses / cycle due to toxicity. SE •Induces proinflammatory cytokines (IL-2, TNFalpha, IFNgamma) •Capillary Leak Syndrome -- Progressive decrease in systemic vascular resistance resulting in decreased intravascular volume –Clinical picture similar to sepsis –Fever, hypotension, peripheral edema, weight gain, pulmonary edema, electrolyte imbalances, arrhythmias, and decreased urinary output. •Other toxicities can include hepatotoxicity, cytopenias, dermatitis, nausea, diarrhea, neuropsychiatric changes, death. |
|
|
Ipilimumab
|
Immune therapy for melanoma and renal cell carcinoma
•Human IgG1 monoclonal antibody against cytotoxic T-lymphocyte antigen-4 (CTLA-4) •Blocks costimulatory signaling in T cells that can inhibit an immune response to tumor •Significantly improves overall survival in metastatic melanoma as compared to usual chemotherapeutic drugs or peptide vaccination –10 versus 6.4 months (previously treated) –Also showed benefit in combination with DTIC versus DTIC alone 1) Co-stimulation via CD28 ligation transduces T-cell activating signals 3) Blocking CTLA-4 ligation enhances T-cell responses 2) CTLA-4 ligation on activated T cells down-regulates T-cell responses 3) Blocking CTLA-4 ligation enhances T-cell responses SE •Autoimmune colitis / diarrhea, which can rarely lead to colon perforation and death in the most extreme cases. •Autoimmune dermatitis •Autoimmune hepatitis (usually transaminitis) •Autoimmune hypophysitis, with decreased levels of thyroid hormone, TSH, cortisol, ACTH, testosterone, FSH, LH •Neurologic side effects •Acquired hemophilia? Unique considerations: •Delayed onset of action •May see new disease prior to response •New criteria for assessing response to autoimmune therapies •Use of steroids to control adverse effects |
|
|
Describe the patient profile, the physical examination findings, diagnostic imaging features, and histopathology of carcinomas of lung, colon, breast, and skin.
|

|
|
|
Langerhans Cell Histiocytosis
(LHC) -cell of origin -MICRO -Name the 3 subtypes |
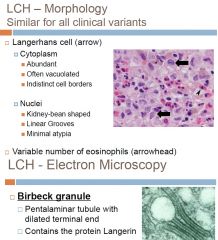
Histiocyte Disorder
Proliferation of immature dendritic cells Originally thought to be derived from skin Langerhans cells (LC) Additional studies have shown LCH to be a proliferation of immature dendritic cells that express the same antigens (including CD1a) as LC Still referred to as LCH and we still refer to the proliferating cell as Langerhans cells Etiology unknown – immunologic dysfunction?? Infection?? Neoplastic or reactive condition?? RARE: ~5 cases per million each year CHILDREN >> adults Multiple clinical variants EM shows Birbeck granules> tennis rackets> role in Ag presentation to T-cells -LCs express HLA-DR, S100, and CD1a CLINICAL PRESENTATION -related to stage of dz -pts with multisystem dz and involvement of BM, liver, and lung are considered high risk 3 Subtypes: 1) Multifocal multisystem (Letterer-Siwe Dz 2) Unifocal and multifocal unisystem (Eosinophilic granuloma) 3) Pulmonary LCH (eosinophilic granuloma of the lung) >>>>see other cards for subtype explanation |
|
|
Multifocal multisystem LCH (Letterer-Siwe Dz)
-what population does it effect? -what organ systems |
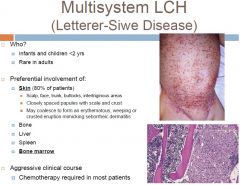
-subtype of Langerhans Cell Histiocytosis
|
|
|
Pulmonary LCH
|
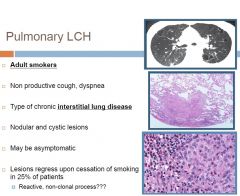
Subtype of Langerhans cell histiocytosis
|
|
|
Intrinsic abnormalities of RBCs (intracorpuscular) causes of anemia
-hereditory vs acquired -clinical problems unique to patients with chonic hemolytic anemias (aplastic crisis vs hemolytic crisis) |
Hereditary
-structural disorders of cell membrane (hereditary spherocytosis) -deficiencies of RBC enzymes (G6P dehydrogenase deficiency) -disorders of Hb synthesis (thalassemias and sickle cell) Acquired -membrane structural defect (paroxysmal nocturnal hemoglobinuria) Clinical problems unique to patients with chronic hemolytic anemia: 1) Aplastic crisis- often due to parvovirus infection -lack of RBC production with continued accelerated destruction can lead to severe prolonged anemia 2) Hemolytic crisis- super-accelerated destruction seen in response to systemic problems such as sepsis |
|
|
Unifocal and Multifocal Unisystem LCH (Eosinophilic granuloma)
|

Subtype of Langerhans Cell histiocytosis
-admixed with eosinophils (prominent), lymphocytes, plasma cells and neutrophils -Hand-Schuller-Christian triad in multifocal dz -prognosis depends on site of involvement, may require systemic chemo |
|
|
Causes of splenomegaly
|
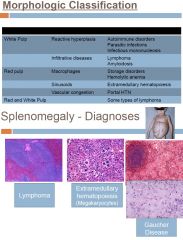
Major manifestation of disorders of the spleen
Clinical presentation depends on the acuteness, nature of underlying disease and the size of spleen LUQ pain, fullness or discomfort Pain referred to left shoulder Early satiety Acute LUQ pain with fever in splenitis or abscess Causes are numerous Hepatic, hematologic, infection or inflammation Spleen enlarges as it performs its normal functions |
|
|
Neoplasms of the spleen
-primary vs secondary -Angiosarcoma vs Hemangioma |
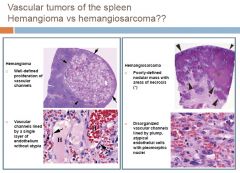
PRIMARY
1)Vascular neoplasms – most common primary neoplasms of spleen a) Hemangioma Most common primary neoplasm of spleen Adults Spontaneous rupture b) Angiosarcoma Most common malignant nonlymphoid neoplasm Adults > 40yrs 2)Lymphoma – as a primary lesion is rare SECONDARY -involvement is rare except in hematolymphoid neoplasms a) Lymphoma – most common malignant neoplasm in spleen b) Metastases Uncommon Carcinomas: lung, stomach, breast, pancreas, liver, colon |
|
|
DiGeorge Syndrome
-classic triad of Sx -what genetic defect? |
Defective development of the pharyngeal pouch system
Deletion of 22q1.2 Large region with many genes Molecular basis of syndrome not fully understood -Congenital Thymus Hypoplasia TRIAD OF SX 1)Cardiac abnormalities 2)Hypoplastic thymus with resultant T-cell immunodeficiency Immunologic dysfunction is variable Total absence of thymus associated with severe immunodeficiency 3)Parathyroid hypoplasia results in hypocalcemia |
|
|
Define renal plasma clearance
|
Renal Plasma Clearance:
•The volume of plasma/min needed to provide the quantity of a substance appearing in the urine each minute •That volume of plasma cleared (freed) of a substance each minute by the kidneys Because inulin is cleared from plasma as the result of glomerular filtration (no reabsorption, synthesis, metabolism or secretion), the renal plasma clearance of inulin (CIN) = GFR The renal plasma clearance can be calculated for ANY substance, providing quantitative information about the renal handling of that substance. |
|
|
Thymic Follicular Hyperplasia
-What is the pathogenesis and clinical features of Myasthenia Gravis? |
-B-cell follicles in thymus
Associated conditions Myasthenia gravis Graves disease SLE Scleroderma RA MYASTHENIA GRAVIS -immune mediated loss of Ach receptors -SX Ocular symptoms (>50% of patients) > Ptosis and/or diplopia Bulbar symptoms (15%) > Dysarthria, dysphagia, fatigable chewing Limb weakness (<5%) |
|
|
List the criteria that a compound must satisfy to be used for measuring Glomerular Filtration Rate (GFR). List some compounds that satisfy these criteria
|
Requirements for a “marker” that will provide quantitative information about GFR:
–Must be free-filterable in glomeruli –Volume of distribution must include plasma –Must be neither reabsorbed nor secreted by the renal tubules –Must be physiologically inert •Non-toxic •No effect on renal function –Must not be synthesized or metabolized by the kidney –Must be readily detected and measured in biological fluid samples -Inulin is freely filtered and all filtered is excreted> use for GFR -amount of inulin excreted per minute is equal to amount filtered per minute |
|
|
Paroxysmal Nocturnal Hemoglobinuria (PNH)
|
Acquired intrinsic RBC membrane defect
•Clinical Presentation Chronic hemolysis (intravascular) Paroxysmal/nocturnal in only 25% Hemosiderinuria Thrombosis of hepatic, portal, or cerebral veins Fatal in 40-50% of cases ∙ Pancytopenia ∙ Complications Myelodysplasia/aplasia/acute leukemia in 5-10% of patients •Hematopoietic stem cell disease due to a somatic mutation of the X-linked gene phosphatidylinositol glycan class A (PIG-A) •Loss of the phosphatidylinositol anchor: certain membrane proteins (including some inhibitors of complement) cannot anchor to the cell surface: CD55, CD59 •RBCs, granulocytes, platelets are abnormal •Median patient survival of 10 years due to potential for serious complications •Diagnosis: flow cytometry (definitive test) for CD55 and CD59 •Diagnosis (historic): increased sensitivity of RBCs to the action of complement (sucrose hemolysis test) •Peripheral smear –Spherocytes –Reticulocytes –Anemia •Bone marrow –Reactive hyperplasia or –Aplastic/hypoplastic |
|
|
Differential of Anterior mediastinal mass?
|
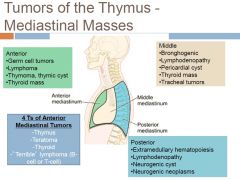
-most common> thymoma
|
|
|
Given data, calculate GFR
|
1.Infuse inulin i.v. until PIN is stable
2.Measure urine volume produced in a known period of time (V) 3.Measure PIN and UIN 4.Calculate GFR GFR= Urine inulin (Uin) x V (flow of urine?) / Plasma inulin (Pin) |
|
|
Name 3 classes of immunohemolytic anemia
|
1) WARM ANTIBODY TYPE (IgG Antibodies Active at 37C)
Primary (idiopathic) Secondary Autoimmune disorders (e.g., SLE) Drugs Lymphoid neoplasms 2) COLD AGGLUTININ TYPE (IgM Antibodies Active Below 37C) Acute (mycoplasma infection, IM) Chronic Idiopathic Lymphoid neoplasms 3) COLD HEMOLYSIN TYPE (IgG Antibodies Active Below 37C) Rare; usually in children after viral infection -aka autoimmune hemolytic anemias (AIHA), Coombs-positive anemia -Coombs = DAT (direct Ab test)> detects Abs on surface of RBCs> AHG (anti-human globulin) used to detect Abs on surface by binding Fc portion |
|
|
Thymoma
-Sx -Morphology -Classification |
Most common anterior mediastinal primary neoplasm in adults
Age: 30-50 yrs Asymptomatic in up to 50% Clinical Presentation -Myastenia gravis -Substernal pain, dyspnea, cough, superior vena cava syndrome -Anemia due to pure red cell aplasia MORPHOLOGY Neoplasm of thymic epithelial cells with variable component of thymocytes Histologic classification based on composition of neoplasm -Epithelial -Lymphocytic Variable cytologic features Benign or malignant CLASSIFICATION Classification based on histology and clinical features: -Noninvasive thymoma -Invasive thymoma -Thymic carcinoma |
|
|
Explain how to decide whether a substance is secreted or reabsorbed
|
It is useful to compare the clearance of a substance to CIN:
If the clearance of a substance is greater than CIN, then there must be net secretion of that substance. If the clearance of a substance is less than CIN, then there must be net reabsorption of that substance (or, alternatively, that substance is not freely filtered). |
|
|
Describe the utility and limitations of plasma creatinine concentration (PCr) as an index of renal function
|
Use of Creatinine Clearance to estimate GFR
•Creatinine is neither synthesized nor metabolized by the kidney •Because creatinine is produced endogenously (a metabolite of creatine phosphate), no i.v. infusion is necessary to determine CCr •Although CCr is slightly greater than CIN (a small amount is secreted), it provides a reasonable estimate of GFR •In reality, CCr is rarely measured in the clinic; rather, PCr is commonly utilized as an index of renal function (normal=1 mg/dl) Cin = Ucr x V/ Pcr |
|
|
Describe the basis for using PAH (para aminohippurate) clearance to estimate renal plasma flow
|
Measurement of Renal Plasma Flow
Renal Plasma Clearance of PAH (CPAH) Renal handling of PAH: 90% of PAH is removed from plasma in a single passage through the kidney ~10% of PAH entering the kidney is not available for secretion CPAH is a measure of plasma flow through parts of the kidney that are effective in removing PAH from plasma — “Effective Renal Plasma Flow” (ERPF) Practical aspects of measuring Effective Renal Plasma Flow 1.Infuse PAH i.v. until PPAH is stable 2.Measure urine volume produced in a known period of time (V) 3.Measure PPAH and UPAH 4.Calculate CPAH Actual Renal Plasma Flow (RPF) •About 90% of PAH is extracted from plasma as it passes through the kidney. –Extraction Ratio of PAH (EPAH) = 0.90 –EPAH = Systemic PPAH – Renal Venous PPAH Systemic PPAH •RPF = CPAH / EPAH |
|
|
Define filtration fraction
|
••Filtration FractionFiltration Fraction(FF) = the fraction (or %) of RPF that is filtered as blood traverses the renal vasculatureFF = GFR / RPFExample: GFR=132 ml plasma/minRPF=623 ml plasma/min••
|
|
|
Given data, calculate
-effective renal plasma flow -renal plasma flow -renal blood flow -filtration fraction |
Renal Blood Flow (RBF)
•RBF = RPF / (1-Hematocrit) “Fractions” in Renal Physiology ••Filtration FractionFiltration Fraction(FF) = the fraction (or %) of RPF that is filtered as blood traverses the renal vasculatureFF = GFR / RPFExample: GFR=132 ml plasma/minRPF=623 ml plasma/min••Fractional ExcretionFractional Excretion(FEQ) = the fraction (or %) of filtered substance (Q) that is excreted in the final urineFEQ= Excreted/min= UQV= CQ/ GFRFiltered/minPQGFRExample: CNa=0.9 ml plasma/minGFR=125 ml plasma/min••Fractional ReabsorptionFractional Reabsorption(FRQ) = the fraction (or %) of filtered substance (Q) that is reabsorbed by the tubulesFRQ= 1 –FEQ |
|
|
Define and classify incidence, prevalence, sensitivity, specificity, positive predictive value, lead time bias, length-time bias and explain how these concepts affect screening practices
|
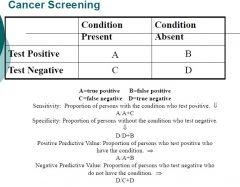
Positive Predictive Value is the most important measurement of the effectiveness of a test.
Positive predictive value varies with the prevalence rate. Screening tests by definition are for conditions that have low prevalence, so tests tend to have high false positive rates. |
|
|
List some types of aplastic/hypoplastic anemias
|
Due to impaired RBC production
–Defective DNA synthesis •ex Megaloblastic anemias (B12, folate deficiency) –Defective hemoglobin synthesis •ex Iron deficiency and thalassemia –Unknown or multiple mechanisms •ex Anemia of chronic disease –Disturbance of stem cell proliferation and/or differentiation •ex Aplastic anemia and pure red cell aplasia |
|
|
Acute Hemolytic Transfusion Rxn
|
1:6000
-intravascular hemolytic occurs within 4 hours of transfusion -hemoglobinemia and hemoglobinuria -fever, flank/back pain, hypotension, pain at transfusion site, anxiety, DIC, vomiting, diarrhea -due to ABO incompatibility> clinical error -fatal in 1:100,000 |
|
|
Transfusion related acute lung injury (TRALI)
|
1:5000
-noncardiogenic pulmonary edema -due to anti-human leukocyte or anti-human neutrophil Abs -respiratory distress within 6 hours of transfusion of plasma containing components> may require mechanical ventilation -fever, hypotension, transient leukopenia, new onset bilateral pulmonary infiltrates on x-ray -does not respond to diuretics or corticosteroids -resolves in 48-96 hours *****MOST COMMON cause of transfusion-related mortality -differential> PE or MI |
|
|
Lead time vs length time bias
|
Lead-time bias: early detection increases time before mortality from the condition
Length bias: early detection increases the number of slowly progressive conditions so severe disease is under-represented. |
|
|
Transfusion associated circulatory overload (TACO)
|
-incidence <1%
-respiratory distress, systolic hypertension, peripheral edema, pulmonary infiltrates on CXR -responds to diuretics -likely underreported -stop transfusion |
|
|
Primary, Secondary, Tertiary prevention.
|
-Primary Prevention: Efforts to prevent the targeted disease (vaccination).
-Secondary Prevention: Efforts to identify and treat asymptomatic persons who have risk factors or pre-clinical disease, but in whom the disease has not become symptomatic (Pap smear, BP checks). -Tertiary Prevention: Efforts to prevent the complications of disease already present (Controlling sugar levels to prevent renal failure). |
|
|
Characteristics, advantages, and limitations of the Gail, Claus, and BRCAPRO risk assessment tools.
|
FOR CANCER RISK ASSESSMENT
1) GAIL MODEL Provides 5-year and lifetime risk estimates based on: – Age – Race (does not distinguish Ashkenazi Jewish) – Age at first live birth or nulliparity – Number of first-degree relatives with a history of BC (maternal history only) – Age at menarche – # of previous breast biopsies – Atypical hyperplasia (ductal and/or lobular) ADVANTAGES •Identifies women who could benefit from preventive interventions; may assist in making clinical decisions (Determination of eligibility for tamoxifen for breast cancer risk reduction…Gail score>1.7) •Incorporates risk factors other than family history (eg, reproductive variables, atypical hyperplasia, history of breast biopsies) •Calculation of breast cancer risk in absence of family history in women •Shows that BC risk increases with age and, therefore, may prompt discussion about the importance of BC screening •Used to counsel and educate women, especially those who overestimate their BC risk LIMITATIONS •Not validated for black, Hispanic, and other ethnic groups •Only solicits family history involving first-degree relatives •May underestimate risk when family history is on father‟s side •Does not take into account age at which relatives developed BC •Effect of number of breast biopsies (without atypical hyperplasia) may cause inflated risk estimates •May underestimate risk for women with demonstrated mutations of the BRCA1 or BRCA2 genes 2) CLAUS MODEL •Risk estimates based solely on family history –incorporates maternal and paternal breast cancer history –first- and second-degree relatives –age at breast cancer diagnosis ADVANTAGES •Maternal and paternal FH •Age of onset of breast cancer •Separate tables to calculate risk of breast cancer based on FH of ovarian cancer +/- breast cancer •Calculation of risk dependent on age of proband LIMITATIONS •requires a set of published tables limited to specific combinations of affected relatives (eg, two first-degree relatives, mother-maternal aunt, etc), and the tables do not include the commonly encountered mother-maternal grandmother pair. •Does not incorporate risk factors other than FH •Currently not validated 3) BRCAPRO Provides estimates for the likelihood of finding either a BRCA1 or BRCA2 mutation in a family: – Bayesian model that incorporates published BRCA1 and BRCA2 mutation frequencies, – breast and ovarian cancer penetrance in mutation carriers – cancer status (affected, unaffected, or unknown) – age of the proband’s first- and second-degree relatives (both maternal and paternal) – Ashkenazi Jewish ethnicity Analysis is based primarily on large, high-penetrance families A validation of this model recently has been published ADVANTAGES •Incorporates both affected and unaffected family members in estimation of carrier probability •incorporates maternal and paternal breast and ovarian cancer history •age at cancer diagnosis, current ages, ages relatives became deceased considered •Ashkenazi Jewish ethnicity taken into consideration •Oophorectomy status and breast cancer receptor status considered LIMITATIONS •Dependent on published estimates of prevalence and penetrance of BRCA1 and BRCA2 •Does not consider more distant family history past 1st and 2nd degree relatives •Does not consider other potential susceptibility genes with features similar to BRCA1 and BRCA2 |
|
|
Aplastic Anemia
|
Normocytic
-BM failure -Pancytopenia (anemia, neutropenia, thrombocytopenia) •Causes –Unknown (65%) –Viral infections •EBV, HIV, CMV •Non ABC hepatitis –Idiosyncratic drug reactions •Chloramphenicol •Streptomycin –Pesticide exposure –Nuclear war/accidents –Fanconi’s anemia (inherited disorder that results in decreased ability to repair DNA defects) •Pathogenesis –Extrinsic: immune-mediated suppression (T-cells reacting against stem cells) –Intrinsic: stem cell abnormality (descendent cells have poor proliferative capacity) •Peripheral smear –Pancytopenia –Normocytic, normochromic anemia –No reticulocytes Bone marrow Markedly decreased or absent hematopoietic cells Fibrosis and fat replacement Clusters of lymphocytes and plasma cells Sx- gradual onset, may be sudden, anemia, bleeding episodes (due to thrombocytopenia), infections (from neutropenia), NO splenomegaly MICRO-hypocellular marrow with marked fat replacement, clusters of lymphocytes and plasma cells Dx- CBC, normochromic, normocytic anemia, no reticulocyte response, empty BM Rx- withdrawal of offending agent (if there is one), antithymocyte globulin and cyclosporine, stem cell transplant |
|
|
Ataxia-Telangectasia
-what gene mutation? |
Autosomal recessive disorder
Neuronal degeneration leads to an ataxic-dyskinetic syndrome beginning in childhood The gene that is mutated in this disorder encodes for ATM: a kinase that senses DNA double-strand breaks caused by ionizing radiation or oxygen radicals and then phosphorylates p53 Mutations in ATM increase sensitivity to x-ray-induced DNA damage ATM carriers may have an increased risk of breast cancer from screening mammography |
|
|
Other forms of Bone Marrow aplasia
-name 3 |
1) Myelophthisic processes
–Space-occupying lesion in the marrow- often seen with extrameduallary hematopoiesis 2) Severe liver disease –Cirrhosis/liver failure –Alcohol 3) Chronic renal insufficiency –Anemia proportional to the severity of uremia> may cause lack of erythropoietin production |
|
|
Overview of genetic mutations and the cancer they cause (Chart)
|
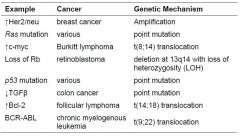
|
|
|
BCR-ABL
-associated with which cancer? -what is it's normal function -how can this be used as a target for therapy? |
-in CML
-hyperproliferation of neoplastic myeloid progenitors -adults 25-60yo -translocation of ABL (chrom 9) and BCR (chrom 22) -ABL- a non-receptor tyrosine kinase that participates in signal transduction pathways of hematopoietic cells -the BCR-ABL protein is constitutively active because it has lost its negative regulatory domain of the tyrosine kinase -Imatinib mesylate (Gleevec) effectively targets this novel fusion protein by interacting with the ATP binding site of ABL and preventing the kinase activity |
|
|
Plummer-Vinson Syndrome
-seen in what scenario? -what is the triad of Sx? |
-seen in Fe deficient anemia
PLUMMER-VINSON SYNDROME -microcytic/hypochromic anemia -atrophic glossitis -esophageal webs |
|
|
ERB B2
-associated with what cancer? -normal function -how do therapies target this? |
-ERB B2 is the human epidermal growth factor receptor-2
-encoded by ERB B2/Her2/Neu oncogene which is amplified in 25% of breast cancers -ERB B2 gene amplification is associated with: 1) more rapid tumor growth 2) poor response to conventional chemo 3) increased risk of recurrence after therapy -Dx tests -IHC of ERB B2 overexpression and FISH to confirm ERB B2 gene amplification -Herceptin (Trastuzumab)- an Ab against ERB B2 is used for targeted therapy |
|
|
Hepcidin
|
-deals with Fe storage and absorption
-increased hepcidin causes: -decreased Fe uptake in duodenum -decreased iron release from macrophages -decreased Fe stores -decreased release of Fe from storage -leads to lower serum Fe? |
|
|
APC and MUTYH
-associated with which cancer? -how are these targets for therapy? |

Colon cancer
-genetic predisposition> develops in younger (<50), multiple polyps, can have two primary colon cancers in same patient -APC and B-catenin are compnents of a signaling pathway activated by ligand binding to WNT (surface receptor) -B-catenin is a transcription factor that regulated cell proliferation and cell adhesion -in the absence of WNT ligands, APC supresses cell proliferation by promoting degradation of B-catenin -deletion or mutation of APC> classic familial adenomatous polyposis (FAP) syndrome> develop hundreds of polyps and 100% lifetime risk of colon cancer -Other mutations in APC (MUTYH gene) cause attenuated FAP> fewer polyps, later age, and 50% chance of colon cancer -other APC mutations> Gardner Syndrome (rare) -Turcot syndrome also part of this group> 2/3 have APC mutations and develop medulloblastomas, other 1/3 have mutations of one of the genes associated with HNPCC and develop glioblastomas -identification of these mutations can be beneficial for early monitoring and prophylactic colectomy |
|
|
Gene Expression Profiling in cancer treatment
|
Gene Chip (microarray)
-used to determine patterns of gene expression in cancer vs normal cells -observed patterns, signatures Use of signatures: 1) classify one tumor type into distinct subgroups (subtypes of diffuse large B-cell lymphomas) 2) predict patient response to therapy by comparing outcomes between groups 3) better understand the biology of the subgroups (germinal center vs activated B cell origin of diffuse B-cell lymphomas) |
|
|
Proteomics in cancer treatment
-3 goals |
-involves searching for proteins that:
1) serve as biomarkers for early dz 2) provide early indicate of response to therapy 3) predict likelihood of relapse after treatment |
|
|
Epigenetic changes in cancer
|
Alterations in DNA, other than in the primary sequence or in the number of gene loci> occur without mutation
Includes DNA methylation and histone acetylation Result in a differences in gene transcription and can contribute to carcinogenesis Cancer: hypermethylation of the promotor region is one mechanism for inactivating tumor suppressors |
|
|
miRNAs (in cancer)
|
Most recently discovered mechanism of gene regulation
Non-coding, ~22 bp long RNAs that influence which mRNAs gets translated> leads to global control of cell growth, differentiation and cell survival Example: the c-myc oncogene controls a miRNA that, in turn, regulates the translation of enzymes used in glutaminolysis |
|
|
Essential Alterations in cancer cells:
-Self-sufficiency in growth signals -give examples |
-normal cells require growth signals to proliferate
-tumor cells generate ther own signals via expression of oncogenes or by causing a normal stromal cell to produce GFs -oncogenes have been discovered at each step of the growth signal circuitry EXAMPLES 1) ERB B2/Her2/neu- breast cancer 2) BCR-ABL fusion protein generated by t(9:22) translocation in CML 3) Ras- multiple cancers 4) c-myc- overexpression due to t(8:14) translocation in Burkitt lymphoma |
|
|
Essential Alterations in cancer cells:
-Insensitivity to anti-growth signals -give examples |
These signals include:
1) soluble growth inhibitors 2) immobilized inhibitors in the ECM 3) inhibitors expressed on the surface of neighboring cells -many of these signals work to regulate cell cycle Cancer develops strategies to ignore these signals: 1) Downregulation of the TGFb receptor 2) Viral inactivation of inhibitors (HPV inhibits Rb tumor suppressor) 3) Overexpression of a cyclin protein 4) loss or mutations in Rb, p53, or APC tumor supressor genes |
|
|
Essential Alterations in cancer cells:
-evasion of apoptosis -give examples |
-normal balance of mitosis and apoptosis
-cancer strategies to evade apoptosis include: 1) overexpression of apoptosis inhibitors 2) mutations in proapoptotic regulators 3) abnormal activity of prosurvival signaling pathways Examples -overexpression of Bcl-2 (antiapoptotic protein) -in lymphoma through the t(14:18) translocation -p53 or PTEN mutations in many types of cancers |
|
|
Essential Alterations in cancer cells:
-limitless replicative potential -give examples |
-most cells reach a period of senescence when replication has reached its limit (shortening telomeres)
-cancers show limitless replicative potential -strategies include: -activation of telomerase -insensitivity to signals generated by shortened telomeres (ex. because of p53 mutations) |
|
|
Essential Alterations in cancer cells:
-sustained angiogenesis -give examples |
-once new tissue is formed, new blood vessel formation is transitory and carefully regulated
-cancer can stimulate angiogenesis once it grows beyond maximum diameter (1-2mm) needed for adequate oxygen and nutrients Ex -VEGF production in glioblastoma multiforme -loss of the anti-angiogenic factor, Von Hippel-Lindau (VHL) protein in renal cell carcinoma |
|
|
Inherited Thrombophilias
-when to suspect this? -chart |
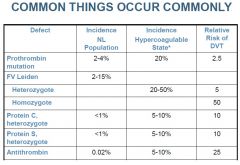
-First thrombosis at a young age (<50 y)
-Recurrent thrombosis -Thrombosis at an unusual site (mesenteric, portal, subclavian veins) -Life-threatening thromboembolism -Family history of thrombosis |
|
|
Abnormalities of the Protein C system
-describe the 3 major abnormalities in this system -what is Factor V Leiden mutation? |
Inherited Thrombophilia
-Protein C and S are Vit K dependent proteins synthesized in the liver -when protein C cleaved by thrombin-thrombomodulin complex it becomes activated (APC) -APC along with its cofactor protein S (PS) inactivate FVa and FVIIIa thus limiting the coagulation cascade 3 major abnormalities: 1) APC resistance- point mutation in FV known as Factor V Leiden mutation and is the most common cause of hereditary thrombosis -FV is resistant to inactivation by APC resulting in loss of one of the inhibitory checkpoints in the cascade -AD inheritance -Dx- clotting based studies, DNA studies 2) Protein C deficiency- -AD -Labs- measure PC activity and Ag levels 3) Protein S deficiency -AD - measure PS activity and Ag levels |
|
|
Prothrombin gene mutation
|
Inherited Thrombophila
-point mutation in prothrombin gene results in increased levels of prothrombin that promote enhanced clot formation DX- molecular studies for the mutation |
|
|
Antithrombin deficiency
|
Inherited Thrombophilia
-AT is an inhibitor of factors XIIa, XIa, IXa, Xa, and IIa (thrombin) -heterozygotes- diminished AT production -homozygotes- lethal in utero DX- measure AT function and Ag level |
|
|
Acute Graft vs Host Dz in HSCT
|
•Donor T-cells infiltrate host tissue and lead to a cytokine storm
•Typically affected organ systems are Skin, GI tract, and Liver •Diagnose by biopsy of affected organ •Traditionally defined as occurring before day 100 after transplantation •Now defined by clinical features •Principal risk factor is HLA mismatch, but may occur despite an HLA-matched donor •Incidence of GVHD reduced by in vitro T-cell depletion of graft before transplantation, but this leads to increased graft rejection and disease relapse •If prophylaxis not provided, serious acute GVHD affects 100% •Prophylaxis for GVH prior to transplant with Methotrexate + either Cyclosporine or Tacrolimus •Treatment after acute GVH occurs is with high dose steroids (2 mg/kg), or anti-thymocyte globulin (ATG) •Steroid Refractory acute GVH has very poor survival due to high risk of opportunistic infection and organ damage from GVH |
|
|
Dissseminated Itravascular Coagulopathy (DIC)
-pathogenesis -acute vs chronic -Sx -Dx/Labs -Rx |
Acquired Thrombotic Disorder
-excessive activation of coagulation results information of microvascular thrombi and secondary consumption of platelets, coag factors, and natural anti-coags -Triggers of DIC include: 1) release of thromboplastic substance into blood (TF or other substance) 2) widespread endothelialinjury -these lead to widespread loss of control of hemostatic and regulatory mechanisms ACUTE DIC -due to massive exposure of TF> overwhelms control and compensatory mechanisms (liver, bone marrow)> do not have sufficient time to respond> consumption of platelets and coag factors (consumptive coagulopathy)> severe bleeding due to microthrombi, tissue ischemia, and microangiopathic hemolytic anemia CHRONIC DIC -aka compensated DIC -occurs when blood is exposed intermittently to small amounts of TF and the compensatory mechs are able to replenish coag factors and platelets> most common cause is malignancy -NOT a primary dz- associated with numerous conditions- obstetric complications, sepsis, malignancy, massive tissue injury CLINICAL FEATURES -Acute DIC- bleeding -microvascular thrombi can cause ischemia and multi-organ failure -Chronic DIC- may be aSx, develop thrombosis or minor bleeding LABS -Acute- thrombocytopenia, hypofibrinogenemia, microangiopathic hemolytic anemia (shistocytes on smear), prolonged PT and aPTT, elevated D-dimer -Chronic- labs variable, platelet count may be mildly decreased, fibrinogen normal or increased, PT and aPTT may be normal RX -TREAT the underlying cause -if serious bleeding> plasma and/or platelet transfusion -heparin for those with thrombotic complications |
|
|
Chronic graft vs Host Dz in HSCT
|
•Affects 40-80% of long term survivors of alloSCT
•Loss of Self Tolerance •Mimics signs of autoimmune disorders, like scleroderma or Sjögren’s syndrome. •Skin, nail, mouth, eye, liver, lungs commonly affected •Can present as keratoconjunctivitis sicca, esophageal stricture, malabsorption, cholestasis, cytopenias, bronchiolitis, and generalized Immunosupression. •Previously defined as occurring >100 days after transplant, but now clinically defined •Treatment with corticosteroids may be needed for two years or longer |
|
|
Other acquired risk factors for thrombosis
|
Major acquired risk factors
Major recent trauma Immobilization Trauma Lupus anticoagulant/APA Pregnancy OCPs Myeloproliferative neoplasms (polycythemia vera, essential thrombocytosis) *****Malignancy -may precede Dx of malignancy Pathogenesis: Cancer patients have multiple reasons to develop thrombosis, consider Virchow’s triad: Stasis – bed rest, vascular compression Endothelial injury –vascular invasion, catheters, chemotherapy Hypercoagulability – substances released from neoplasm may be procoagulant |
|
|
Sinusoidal Obstruction/Hepatic veno-occlusive dz in HSCT
|
•Clinical triad of weight gain +/- ascites, tender hepatomegaly, and jaundice
•Caused by conditioning regimen toxicity, and results in damage to endothelial cells, sinusoids and hepatocytes •Risk factors •Patients with liver disease prior to transplant •Heavy chemotherapy pre-treatment prior to transplant •Prolonged, elevated Busulfan levels •>12 Gray total body irradiation •Diagnosed by liver ultrasound and biopsy •Occurs 8-10 days after prep regimen. High mortality rate •New drug Defibrotide (adenosine receptor agonist) has response rate of 36% |
|
|
Pulmonary toxicity in HSCT
|
•Occurs within 4 months of transplant
•Mortality rates of 60% •Differential Diagnosis •Idiopathic pneumonia syndrome •Diffuse alveolar hemorrhage •Obstructive Airway Disease (Bronchiolitis obliterans) •Treatment of above diseases is mostly supportive, but may include high dose steroids •Differentiate from “Engraftment Syndrome” •During neutrophil recovery •High fever, fluid retention, and diffuse pulmonary infiltrates |
|
|
Heparin Induced Thrombocytopenia (HIT)
-type I vs type II -Sx -Labs/Dx -Rx |
-TCP due to decreased platelet survival
2 types: 1) Type I- TCP starts immediately after heparin admin, likely due to direct platelet-aggregating effects of heparin> usually clinically insignificant 2) Type II- potentially devastating syndrome resulting form immune-mediated platelet activation -more common in adults than children, occurs 5-14 days after starting heparin, less common than type I PATHOGENESIS (type II) -IgG to heparin platelet factor 4 complex> platelet activation and aggregation> promotes thrombosis even in TCP> platelet aggregates removed from circulation leading to TCP SX -DVT and PE are main manifestations -severe TCP with bleeding is rare> major problem is thrombosis -occurs more frequently in pts with unfractionated heparin (vs LMWH) and in surgical patients (vs medical) LABS -platelets <150k or 40-50% of pre-heparin count (it may be normal!!!) -HIT Ab can be detected by immunoassay or functional studies that detect platelet activation in presence of heparin Rx -discontinue heparin -give alternate drug> direct thrombin inhibitor -do not use LMWH> can cross react -avoid warfarin> reduces protein C which can cause more thrombosis |
|
|
List the major causes of Lymphoid neoplasia
(5 categories) |
1) Chromosomal translocations and oncogenes
–Immunoglobulin genes in B cells and T cell receptors in T cells rely on physiologic breakage, recombination and somatic mutations to produce antigen receptor diversity –Oncogenes can easily be translocated to these regions, then over expressed by the Ig and TCR promoters, eg. BCL2 and MYC 2) Inherited genetic factors: disorders of genomic instability, Down syndrome, NF type I 3) Viruses: –EBV: Burkitt’s, Hodgkin’s, and post-transplant 4) Chronic Immune Stimulation •Helicobacter pylori and MALT lymphoma •Gluten sensitive enteropathy and intestinal lymphoma •HIV and B cell lymphoma 5) Iatrogenic –Radiotherapy, Chemotherapy |
|
|
Uremia
-path -sx -labs -rx |
Acquired Platelet function disorder
Path- impaired platelet adhesion and aggregation (mech not understood) Sx- mucocutaneous bleeding Labs- impaired platelet aggregation Rx- correct anemia, dialysis, estrogen treatment (mech unknown) |
|
|
Immunodeficiency associated large B cell lymphoma
|
•Clinical scenarios
–acquired immunodeficiencies •HIV, drug-induced, following solid organ transplant –inherited immunodeficiencies •Often involve the brain (HIV+ cases) •Often latently infected with EBV •May regress with restoration of T cell immunity •Treatment involves decreasing immunosuppression and/or chemotherapy |
|
|
Von Willebrand Dz
-Path -Sx -Labs -Rx |
Inherited Coag Factor Deficiency
PATH -AD, rarely AR -decreased vWF or impaired function -vWF is a large multimeric glycoprotein synthesized by endothelial cells and megakaryocytes with 2 main functions: 1) carrier molecule for factor VIII 2) promotes platelet adhesion to subendothelial collagen through the GPIb receptor Sx -most common inherited bleeding disorder -hallmark is mucocutaneous bleeding (epistaxis, menorrhagia, ecchymosis) but can also present with spontaneous soft tissue bleeding from minor trauma or wound bleeding LABS -abnormal platelet function testing, normal platelet count -prolonged aPTT (due to decrease in factor VIII) and decreased vWF activity -Electrophoresis- abnormal or decreased vWFmultimers -classified by multimeric structure of vWF, vWF function and levels required for direct therapy RX -DDVAP (vasopressin)- releases vWF and factor VIII from endothelial cells -vWF concentrates |
|
|
Vit K deficiency
-path -Sx -Labs -Rx |
Acquired coag factor deficiency
Vitamin K is an active coenzyme in the carboxylation of glutamic acid residues required for the function of factors II, VII, IX, and X. Causes of deficiency: malnutrition, impaired intestinal absorption, antimicrobial inhibition of the gut flora that provide vitamin K. Clinical features: easy bruising, GI bleeding, hematomas Laboratory: prolonged PT early, both PT and PTT will be prolonged in severe deficiency Treatment: Vitamin K; FFP in severe bleeding |
|
|
Patient Workup for Lymphoma
-Hx -Px -Labs -Radiology -Pathology |
•History:
–Presence, timing, course of lymphadeopathy, masses, rashes –Systemic symptoms (fever, weight loss, night sweats) –Questions related to altered CBC values (fever, infection, bruising, fatigue) –Extra-nodal masses (early satiety) –Risk factors: immune suppression, autoimmune diseases, infections, radiation exposure, family history •Physical Exam: –All peripheral lymph node areas: •Anterior and posterior cervical •Supraclavicular •Axillary •Inguinal •Femoral –Other lymphoid areas •Tonsils •Spleen –Signs of bone marrow involvement •Pallor, fever, petechiae –Extra-nodal organ extension •Lymphoid pressure on veins (limb swelling) –Testicular exam •Blood: –Complete blood count –Comprehensive metabolic panel (liver and kidney function) –Lactate dehydrogenase (NHL) or sedimentation rate (HL) –If suspect tumor lysis: calcium, phosphorus, uric acid –HIV test if suspecting HL or high-grade lymphoma •Radiology: –CT chest, abdomen, pelvis –CT neck if involved (clinical exam is usually sufficient) –Positron emission tomography (PET) scan in select cases (particularly diffuse large B-cell lymphoma and HL) •Pathology: –Bone marrow biopsy (and aspirate, particularly if suspect lymphoid leukemia) –Lumbar tap needs to be performed if suspect highly aggressive lymphoma or some subtypes of aggressive lymphoma |
|
|
Severe Liver Dz (in regards to coag factor deficiency)
-path -Sx -Labs -Rx |
Acquired coag factor deficiency
Hemostatic defects are numerous: o Decreased synthesis of clotting factors o Thrombocytopenia due to splenomegaly, platelet antibodies and/or decreased marrow production Clinical features: These patients bleed from extensive impairment of their hemostatic mechanisms in addition to being at high risk from bleeding from esophageal varices, gastritis, and peptic ulcers. Laboratory: prolongation of PT and aPTT, thrombocytopenia Treatment: Fresh frozen plasma, platelets, and/or vitamin K. |
|
|
Treat Indolent Lymphoma
|
•The paradigm is watchful waiting until treatment criteria are met
•Treatment criteria based on signs & symptoms: –Cytopenias: thrombocytopenia or neutropenia –Leukemic phase (circulating lymphoma cells in the peripheral blood) –Systemic symptoms –Marked splenomegaly, pleural effusion, ascites, or compressive symptoms –Single lymphoma site > 7 cm in diameter –At least 3 lymphoma sites > 3 cm in diameter •Involved field radiation therapy – in early stage •Rituximab – anti CD20 antibody, (CD20 is a typical cell surface protein on B-cell lymphomas) –With or without various chemotherapy regimens |
|
|
Treat Aggressive Lymphoma
|
•Rituxan with combination chemotherapy including steroids are the current standard of care
•Early stage: 3-4 cycles of combination chemo , every 3 weeks, followed by involved field radiation therapy •Advanced stage: 6-8 cycles, radiation omitted since lymphoma is so wide spread •Highly Aggressive: Treated as acute leukemia including CNS prophylaxis |
|
|
Von Hippel Lindau Dz
|
Von Hippel-Lindau Disease
Autosomal dominant disease Frequency: 1:30,000-40,000 Pathology: capillary hemangioblastomas in the CNS and retina cysts in the pancreas, liver and kidney predisposition to developing renal cell carcinoma mutations disrupt the turnover of VHL target proteins HIF1α under normoxia: -hydroxylated -is bound by the Von Hippel-Lindau protein & degraded -is a short-lived protein HIF1α under hypoxia: -not hydroxylated -is a stable transcription factor -gene targets include VEGF (vascular endothelial growth factor) |
|
|
Treat Hodgkins Lymphoma
|
•Combination chemotherapy
•Early stage: 2-4 cycles of chemotherapy, followed by involved field radiation therapy •Advanced stage: 6-8 cycles of chemotherapy without the radiation |
|
|
Porphyria
|
• Rare group of blood disorders caused by an alteration
in one of the enzymes responsible for the biosynthesis of heme • When one of the enzymes responsible for heme production is not functioning, the heme precursors known as porphyrins accumulate • Can cause life‐threatening attacks of neurovisceral symptoms that mimic many other acute medical and psychiatric conditions • Lack of clinical recognition often delays effective treatment • Three main types (eight total variations) – Acute intermittent porphyria – Porphyria cutanea tarda – Congenital porphyria Acute intermittent porphyria • Most important of the three • Clinically distinguishable from the other two forms by the dominance of gastrointestinal and neurological symptoms and the absence of skin photosensitivity • Attacks rare before puberty and after menopause • When the acute attack has lasted a few days the patient often develops a peripheral neuritis which may cause total paralysis which often leads to death Porphyria cutanea tarda • Uroporphyrinogen decarboxylase deficiency • Light sensitive dermatitis • Autosomal dominant inheritance Congenital porphyria • Starts early in childhood • See staining of the bone, anemia, and splenomegaly • Considered erythropoietic in origin • Clinically see skin photosensitivity, hypertrichosis, reddish brown staining of primary teeth • Recessive disorder due to single gene defect • male sex Diagnosis • Acute porphyria should be p p y considered in any patient with unexplained abdominal pain • Often an acute abdominal emergency is diagnosed and the patient is operated on • Markedly increased urinary porphobilinogen level • Urine delta‐aminolevulinic acid (ALA) can also be elevated • Can also have excess porphobilin which is responsible for the reddish brown color of the urine • Often see hyponatremia, hypochloremia and azotemia • Radiographs usually negative • Often a diagnosis of exclusion 22 Treatment • Intravenous hemin therapy most effective • Intravenous glucose therapy alone is appropriate only for mild attacks (mild pain, no paresis or hyponatremia) • Eliminate any precipitating factors • Appropriate supportive and symptomatic therapy should be initiated |
|
|
SE of therapies for lymphoma (chemo)
|
•Chemotherapy (many others besides those listed)
–Bone marrow suppression (chemo) •Thrombcytopenia, leukopenia, neutropenia with associated infections and bleeding –Doxorubicin: Congestive heart failure –Alkylating agents: increased risk of myelodysplasia or AML –Altered fertility –Bleomycin: lung fibrosis •Transfusion associated risks (hemolysis, infection) •Biologics (monoclonal antibodies): immunosuppression, allergic reactions •Radiation: inflammation, fibrosis, increased long term risk of breast cancer and other solid tumors, hypothyroidism |
|
|
MSH2
|
MSH2 and Colon Cancer
MSH2 encodes a protein involved in mismatch repair Hereditary nonpolyposis colorectal cancer (HNPCC) syndrome is caused by mutations in MSH2 Homozygous loss of MSH2 increase gene mutations rates by 1000-fold and leads to genomic instability HNPCC patients develop colon cancer at a younger age (<50) than unaffected individuals |